摘 要:以亚麻屑为原料,(NH4)2HPO4-H3PO4为复合活化剂制备出亚麻基活性炭吸附剂。采用正交实验法,选取复合活化剂(NH)2HPO4与H3PO4质量比、浸渍时间、微波功率及辐射时间为影响因素,以碘吸附值、亚甲基蓝吸附值及产率为评价指标确定最佳制备工艺,通过热重、X射线光电子能谱及N2吸附-脱附测定等手段对最优产物进行结构分析。研究结果表明,最佳制备条件为(NH)2HPO4与H3PO4质量比为1:3,浸渍时间为60min,微波功率400W和辐射时间10min,该条件下制备的活性炭碘吸附值为1142.486mg·g-1,亚甲基蓝吸附值为163mg·g-1,产物得率为40.21%。经过(NH)2HPO4-H3PO4浸渍后,能有效降低原料热解的炭化温度,且能提高产物得率。产物比表面积为1397.86m2·g-1,平均孔径为2.74nm,属于中孔型吸附剂,同时存在微孔结构;其表面存在C-O、O-C=O、-OH和C=O等含氧基团。
关键词:亚麻屑;活性炭吸附剂;复合活化
活性炭吸附剂的制备原料大致可以分为合成纤维和生物质基材料两大类[1]。相对于传统的合成纤维而言,生物质基材料来源广泛,具有可再生性,还具备良好的生物可降解性,在环境保护领域有着广阔的应用前景[2-5]。
我国是世界上亚麻制品的主要生产国和消费国之一。在亚麻纤维加工过程中会产生大量的亚麻屑,每年产量高达数百万吨,主要用于制造板材和造纸,其利用率非常低。因此,寻求亚麻屑资源的高效利用至关重要。作为一种富含有机组分的生物质材料,其具备制成生物质基活性炭吸附剂的条件[6]。利用亚麻屑制备活性炭吸附剂,可以拓宽活性炭制备的原料种类,优化废弃资源配置,促进资源的可持续利用。
本文以亚麻屑(Flax Shive,FS)为原料,采用(NH4)2HPO4-H3PO4活化法制备亚麻基活性炭吸附剂(Flax Activated Carbon Adsorbent,F-ACA),以碘吸附值(QI2)、亚甲基蓝吸附值(QMB)及产率(Y)为主要评价指标确定最佳制备工艺,并通过XPS、热重分析及N2吸附-脱附等手段对F-ACA结构进行表征分析,以期为亚麻屑资源的资源化利用提供了新的思路。
1 实验部分
1.1 主要材料与仪器
材料:亚麻屑,经多次浸泡洗涤、去除表面杂质,经干燥、恒重后备用。
仪器:BS223S型电子天平;DZF-7050型真空干燥箱;SK2200LHC型数控超声波清洗器;SHA2型冷冻水浴恒温振荡器;ML08S-2B型微波反应器(南京汇研微波系统有限公司);7600-1CRT紫外可见分光光度计(上海菁华科技仪器有限公司);STA449F3同步热分析仪(德国耐驰公司);ESCALAB250XiXPS光电子能谱仪(美国赛默飞公司);ASAP2020全自动比表面积及微孔吸附分析仪(麦克默瑞提克(上海)仪器有限公司)。
1.2 F-ACA的制备

图1F-ACA的制备装置简图
(1:氮气瓶;2:固定夹;3:石英烧瓶;4:微波辐射装置;5:安全瓶;6:缓冲瓶;7:洗瓶)
称取8g左右的FS与40ml一定比例的(NH4)2HPO4(30%)、H3PO4(20%)溶液混合,在53kHz,100W的超声波下浸渍,之后真空干燥,将干燥好的FS转移到微波反应器中,通入氮气,在一定功率下辐射至规定时间,待降至室温后取出,得到F-ACA粗产品。用蒸馏水反复清洗至滤液pH为6-7,烘干,备用。其制备工艺装置如图1所示。
1.3 性能测试与表征
亚麻基活性炭吸附剂样品产率是为活化后最终产物的质量与恒重亚麻屑原料质量比(%);采用GB/T12496.10-1999测试亚甲基蓝吸附值;GB/T1246.8-2015测试碘吸附值;
采用热重分析对比FS浸渍前后的热解过程。样品干燥2小时后测定,氮气气流速度为10mL·min-1,热重升温速率15℃/min,温度范围为30-800℃;
采用同步热分析仪法测定N2吸附-脱附等温线。干燥的F-ACA样品在350℃,N2的保护下脱气处理4h后测定,通过BET法和BJH法计算样品的比表面积和孔径分布;
采用XPS对FS和F-ACA样品进行表面结构分析。真空度为2×10-9Pa,分辨率为0.5eV,扫描范围0-1400eV,使用Avangage软件进行分峰拟合。
1.4 制备工艺的正交实验分析
根据前期试验探索的结果分析,选取复合活化剂(NH)2HPO4与H3PO4比例(质量比)、浸渍时间、微波功率及辐射时间等作为F-ACA制备工艺中吸附性能影响较大的因素,以碘吸附值(QI2)、亚甲基蓝吸附值(QMB)、产率(Y)作为评价吸附性能的考察指标,进行4因素3水平的正交试验。
由于本试验是多指标正交,因此,采用综合评分法对试验结果进行分析。根据多个评价指标的重要程度,将其转化为隶属度,用隶属度代表分数,再求出综合分。隶属度计算方法如公式(1)所示

(1)综合分=QI2隶属度×0.2+QMB隶属度×0.6+Y隶属度×0.2
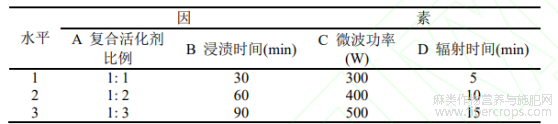
采用L9(34)正交实验确定制备的最佳工艺,试验因素水平表如表1所示。
表1试验因素水平表
2 结果与讨论
2.1 制备工艺的优化
表2正交试验结果
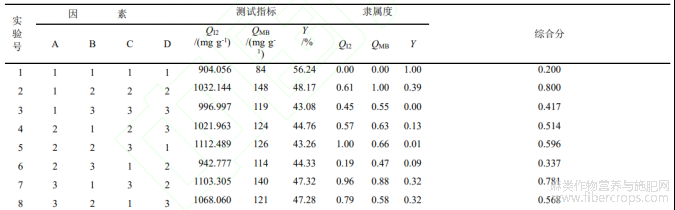

表3产率、碘吸附值和亚甲基蓝吸附值极差分析
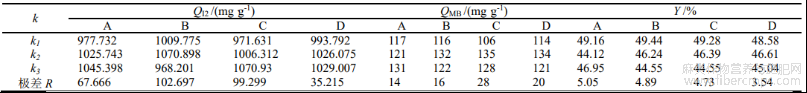
由表2可知,不同工艺制备的活性炭的吸附指标基本上达到了粉状活性炭的国家标准(QI2大于900mg·g-1,QMB大于100mg·g-1),不同制备工艺下的产率基本上在40%-50%之间,相差不大,说明质量损失率在50%-60%之间。在不考虑各单因素交互作用的基础上,由表2和表3的极差分析R可知,4个因素对QI2的影响次序为:浸渍时间>微波功率>(NH)2HPO4与H3PO4质量比>辐射时间;4个因素对QMB的影响次序为:微波功率>辐射时间>浸渍时间>(NH)2HPO4与H3PO4质量比;4个因素对综合评价分影响的显著性由大到小依次为微波功率>浸渍时间>辐射时间>(NH)2HPO4与H3PO4质量比。研究结果发现,微波功率均是重要的影响因素。分析是因为微波功率与活化温度有很大关系,直接影响到活化反应的效能和产物的孔隙结构的变化[7]。表2的试验结果显示,最优工艺方案为A3B2C2D2,即:(NH)2HPO4与H3PO4质量比为1:3,浸渍时间为60min,微波功率和辐射时间分别为400W和10min,在此最佳条件下制备的活性炭碘吸附平均值为1142.486mg·g-1,亚甲基蓝吸附平均值为163mg·g-1,产物得率为40.21%,吸附性能优于正交的9组试验,仅仅产率略有下降,分析是因为活化程度的提高能加速炭化物的失重程度。
2.2 性能表征
2.2.1 孔结构分析
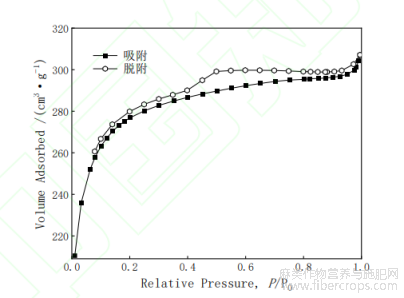
图2F-ACAN2吸附-脱附等温线
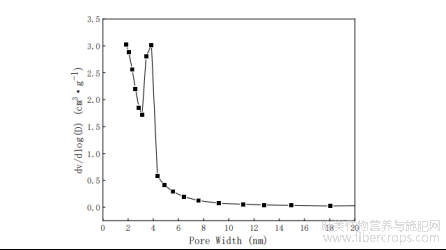
图3F-ACA的孔径分布
F-ACA的N2吸附-脱附曲线如图2所示。按照IUPAC的划分,F-ACA的吸附等温线类型为IV型,曲线中出现明显的H4型迟滞环,表明样品以中孔结构为主[8]。对于吸附支曲线来说,当相对压力P/P0=0-0.2时,随着相对压力的增加N2吸附量急剧升高,这表明F-ACA的微孔发挥主导作用[9],并被迅速填充;当相对压力在P/P0=0.2-0.8之间时,吸附速率变慢,曲线趋于平缓,有吸附平台出现,说明F-ACA具有稳定的结构;P/P0高于0.8后,吸附量随着相对压力的增大渐渐接近饱和值,吸附量略有增加。对于脱附支曲线来说,当P/P0处于0.5-1.0之间时,脱附速率平稳;当P/P0低于0.5时,脱附速率变快,表明F-ACA中大于N2分子尺寸(0.304nm)的孔径起主导作用。
F-ACA孔径分布曲线见图3。根据图2可知,F-ACA孔径主要分布2-4nm范围内,中孔分布广泛,数量较多。通过BJH法计算F-ACA平均孔径为2.74nm,表明样品以中孔结构为主。
根据表4中BET比表面积相关参数可知,单点SBET与多点SBET相差不大,表明F-ACA有较强的吸附能力;BET比表面积为1397.86m2·g-1,中孔比表面积约为总比表面积的75.41%,表明F-ACA中有较多中孔,这与图2曲线得出的结论一致。因此,F-ACA中含有丰富的中微孔结构。
表4F-ACA的比表面积参数
![]()
2.2.2XPS分析
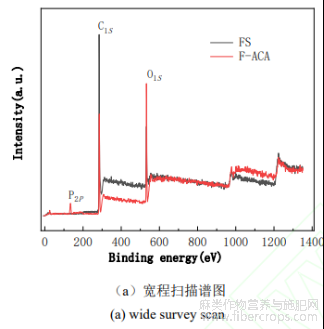
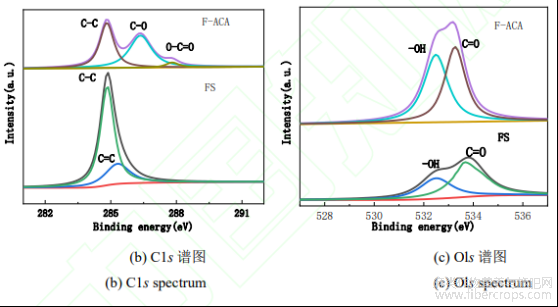
图4FS和F-ACA的XPS图谱
采用XPS能谱对FS和F-ACA的表面结构进行分析,其结果如图4所示。由图4(a)全谱可知,FS和F-ACA样品在285.1Ev和532.1eV处均有较强的特征光电子峰,F-ACA样品在134.1ev处还出现较弱的P2p特征峰,这表明FS活化后,不仅存在C、O元素,还有P元素掺杂并进入炭化物的孔结构中。对FS和F-ACA样品的C1s、Ols进行分峰拟合,从图4(b)和图4(c)可以看出,FS和F-ACA样品C1s谱图284.8、285.4、286.4、287.8eV处的特征峰分别归属于C-C、C=C、C-O、O-C=O等基团[10,11],样品Ols谱图532.5、533.5eV处峰分别归属于-OH和C=O基团[12]。通过XPS能谱分析结果可知,F-ACA的C1s质量分数从FS的83.41%降至69.94%,而Ols的质量分数由FS的13.13%升至23.23%。F-ACA样品分峰图中无C=C特征峰的出现,应是微波热解时碳碳双键被破坏导致的;-OH和C=O的质量分数比例有所增加,这与Ols质量分数的变化相吻合。综合以上分析,该制备F-ACA表面存在较丰富的C-O、O-C=O、-OH和C=O等含氧基团,这增加了F-ACA的亲水性和表面活性位点,有利于该活性炭吸附性能的提高。
2.2.3 热解历程分析
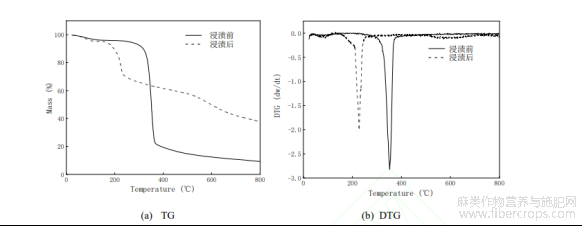
图5浸渍前后FS样品的TG和DTG
图5是复合活化剂浸渍前后样品的TG-DTG曲线对比。由图5可知,浸渍前后FS样品的热解过程经历了三个阶段:第一阶段的失重发生在100℃以下,主要是水分的去除,但浸渍后样品DTG曲线显示,在80℃左右出现了较弱的失重峰,可能是浸渍用复合活化剂具有一定亲水性的缘故。第二阶段是200-450℃的炭化过程,该过程属于快速热解阶段,浸渍前后均出现了较明显的重量变化,炭基结构被破坏,化学键发生断裂。由图5(a)可以看出,浸渍前约有80%的失重率,浸渍后失重率明显降低,说明炭基材料的热解过程被复合活化剂改变,H3PO4能促进FS的纤维素和半纤维素的水解,经过羟基缩合作用形成磷酸酯[13],同时,(NH4)2HPO4热解可产生聚磷酸铵[14],这些物质能抑制焦油生成,对碳骨架具有一定的保护作用,使成炭率升高[15,16];从图5(b)的DTG曲线还发现,浸渍前样品在350℃左右有一个较强的失重峰,而浸渍后样品在225℃左右出现一个较强的失重峰,说明复合活化剂的浸渍可以降低FS样品的炭化温度,分析原因主要是(NH4)2HPO4在170-250℃之间将分解成氨气和含磷的化合物[17,18],同时,H3PO4在此温度范围内也会失水发生环化和缩合作用,加速样品的键断裂和链交联,不仅有效提高炭化物的产率,还降低了炭结构形成的温度,促进稳定结构碳骨架的生成;第三阶段是大于450℃的过程,浸渍前的失重曲线趋于平缓,可表明在此区间内有相对稳定的炭化产物生成。但浸渍后的失重曲线出现了一定程度的下降,可能是生成的五氧化二磷(分解温度560℃左右)分解以及其它一些单质如含磷蒸气造成的[19]。综合分析,采用复合活化剂浸渍法,不仅降低了炭基材料的炭化温度,而且提高了产物的得率。
3 结论
以亚麻屑为原料,采用多指标综合评分-正交试验法制备亚麻基活性炭吸附剂。制备F-ACA的最佳工艺为:(NH)2HPO4与H3PO4质量比为1:3,浸渍时间为60min,微波功率400W和辐射时间10min,该条件下制备的活性炭吸附剂碘吸附平均值为1142.486mg·g-1,亚甲基蓝吸附平均值为163mg·g-1,产物得率为40.21%。各因素对综合评价分影响排序为微波功率>浸渍时间>辐射时间>(NH)2HPO4与H3PO4质量比。
F-ACA具有H4型迟滞环,属于IV型等温线,比表面积为1397.86m2·g-1,中孔占比75.41%,平均孔径为2.74nm,是以中孔为主,同时存在微孔结构的吸附剂。F-ACA表面存在大量的C-O、O-C=O、-OH和C=O等含氧基团活性位点,具有一定的亲水性。热解历程分析表明,经过(NH)2HPO4-H3PO4复合活化剂浸渍后,FS热解的炭化温度得以降低,并有效提高了产物的得率。
参考文献
[1]曹向禹,田俊阳,李维鑫.活性炭纤维制备技术的研究进展[J].应用化工,2020,49(04):1020-1024.
[2]FOO K Y,HAMEED B H.Microwave-assisted preparation of oil palm fiber activated carbon for methylene blue adsorption[J].Chemical Engineering Journal,2011,166(2):792-795.
[3]LI J,NG D,SONG P,et al.Preparation and characterization of high-surface-area activated carbon fibers from silkworm cocoon waste for congo red adsorption[J].Biomass and Bioenergy,2015,75:189-200.
[4]YANG X,ZHAO S,ZHANG Z,et al.Pore structure regulation of hierarchical porous carbon derived from coal tarpitch via pre-oxidation strategy for high-performance supercapacitor [J].Journal of Colloid and Interface Science,2022,614:298-309
[5]SUN Z W,DUAN X H,SRINIVASAKANNAN C,et al.Preparation,optimization and characterization of carbon fibers adsorbent from cotton by microwave induced ZnCl2 activation[J].Science of Advanced Materials,2018,10(5):724-733.
[6]张丽,刘梁森,邱冠雄.废弃纺织材料回收利用的研究进展[J].纺织学报,2013,34(04):153-160.
[7]田叶顺,任文,王国袖,等.微波加热CO2活化法制备生物质活性炭及其脱硫性能研究[J].化工学报,2020,71(12):5774-5784.
[8]OH G H,PARK C R.Preparation and characteristics of rice-straw-based porous carbons with high adsorption capacity[J].Fuel,2002,81(3):327?336.
[9]李欢欢,李海红,吴丹萍.KOH活化法优化制备辣椒秸秆基活性炭及其性能表征[J].化学研究与应用,2022,34(9):2149-2156
[10]蒋小雪,李翠清,张伟,等.改性酚醛树脂基活性炭对二氯甲烷的吸附[J].化工环保,2023,43(6):829-837
[11]吴连永,张大琴,贾志刚,等.木焦油基活性炭的制备及其对亚甲基蓝的吸附性能[J].精细化工,2023,40(1):177-184,232
[12]PI X X,QU Z B,SUN F,et al.Catalytic activation preparation of nitrogen-doped hierarchical porous bio-char for efficient adsorption of dichloromethane and toluene[J].Journal of Analytical and Applied Pyrolysis,2021,156:105150-105162
[13]邓锋,解强,刘德钱,等.2~5nm孔集中分布泥炭基中孔活性炭的制备[J].化工学报,2019,70(11):4457-4468
[14]TANG Y B,LIU Q,CHEN F Y.Preparation and characterization of activated carbon from waste ramulus mori[J].Chemical Engineering Journal,2012,203:19-24.
[15]ZHOU X,WANG P L,ZHANG Y G,et al.From waste cotton linter:a renewable environment-friendly biomass based carbon fibers preparation[J].ACS Sustainable Chemistry & Engineering,2016,4(10):5585-5593
[16]LIN G F,WU K K,Huang B.Effects of small amounts of phosphoric acid as additive in the preparation of microporous activated carbons[J].Materials Science,2018,24(4):362-366
[17]STATHEROPOULOS M,KYRIAKOU S A.Quantitive thermogravimetric mass spectrometric analysis for monitoring the effects of fire retardants on cellulose pyrolysis[J].Analytica Chimica Acta,2000,409(1-2):203-214
[18]MARCILLA A, BELTRAN M I,GóMEZ-SIURANA A,et al.TGA/FTIR study of the pyrolysis of diammonium hydrogen phosphate-tobacco mixtures[J].Journal of Analytical and Applied Pyrolysis,2015,112:48-55
[19]赵茂爽,冯莉.磷酸氢二铵法制备高产率褐煤基活性炭[J].中国矿业大学学报,2014,43(02)314-318.
文章摘自:曹向禹,张隆新,刘国煜,等.亚麻屑制备活性炭吸附剂及其性能表征[J/OL].应用化工,1-8[2026-05-24].
