摘 要:本发明提供了一种全生物降解的苎麻改性材料及其制备方法,属于全生物降解材料技术领域。所述苎麻改性材料包括以下原料:苎麻粉、聚己二酸/对苯二甲酸丁二酯、聚乳酸、偶联剂、无水乙醇和环戊油。制备方法为:将苎麻粉和偶联剂混合后在搅拌状态下进行一次混炼,然后加入聚乳酸和聚己二酸/对苯二甲酸丁二酯进行二次混炼,最后加入无水乙醇和环戊油经过三次混炼后得到混合料;将混合料经过挤出和注射成型后得到苎麻改性材料。本发明使苎麻中的纤维素分子能够完美地与基体树脂实现两相界面的共混共熔,可实现纤维素的高质量份数的填充,使苎麻改性材料性能稳定,满足不同应用场景的要求。
权利要求书
1.一种全生物降解的苎麻改性材料,其特征在于,所述苎麻改性材料包括以下原料:苎麻粉、聚己二酸/对苯二甲酸丁二酯、聚乳酸、偶联剂、无水乙醇和环戊油。
2.根据权利要求1所述的全生物降解的苎麻改性材料,其特征在于,所述聚己二酸/对苯二甲酸丁二酯和聚乳酸的添加量为原料总质量的70wt.%-87wt.%;
所述苎麻粉的添加量为原料总质量的10wt.%-30wt.%;
所述偶联剂的添加量为原料总质量的0.5wt.%-0.8wt.%;
所述无水乙醇的添加量为原料总质量的2.0wt.%-3.0wt.%;
所述环戊油的添加量为原料总质量的0.5wt.%-2.0wt.%。
3.根据权利要求2所述的全生物降解的苎麻改性材料,其特征在于,所述聚己二酸/对苯二甲酸丁二酯和聚乳酸的质量比为0.6-0.8。
4.根据权利要求2所述的全生物降解的苎麻改性材料,其特征在于,所述苎麻粉的粒度为500-700目。
5.根据权利要求2所述的全生物降解的苎麻改性材料,其特征在于,所述偶联剂为硅烷偶联剂或铝酸化合物。
6.一种如权利要求1-5任一项所述的全生物降解的苎麻改性材料的制备方法,其特征在于,包括以下步骤:
(1)将所述苎麻粉和偶联剂混合后在搅拌状态下进行一次混炼,然后加入所述聚己二酸/对苯二甲酸丁二酯和聚乳酸进行二次混炼,最后加入无水乙醇和环戊油经过三次混炼后得到混合料;
(2)将所述混合料经过挤出和注射成型后得到所述苎麻改性材料。
7.根据权利要求6所述的全生物降解的苎麻改性材料的制备方法,其特征在于,步骤(1)中所述搅拌的转速为20-40rpm;
所述一次混炼的时间为15-30min;
所述二次混炼的时间为5-8min;
所述三次混炼的时间为3-5min。
8.根据权利要求6所述的全生物降解的苎麻改性材料的制备方法,其特征在于,步骤(2)中所述挤出过程设置各区温度为:170℃、170℃、170℃、175℃、175℃、175℃、175℃、175℃、175℃,转速为150rpm,主喂料频率为4.0Hz,主机电流为13A。
9.根据权利要求6所述的全生物降解的苎麻改性材料的制备方法,其特征在于,步骤(2)中所述注射成型过程设置各区温度为180℃、190℃、200℃,注射压力为125bar,射胶流量为60%。
技术领域
本发明属于全生物降解材料技术领域,尤其涉及一种全生物降解的苎麻改性材料及其制备方法。
背景技术
近年来,塑料的使用量日益增大,塑料制品在我们日常生活中随处可见,为人们的生活提供了极大的方便,但随着塑料产量的极速增加,废弃塑料的后处理及造成的环境污染越来越受到各国的广泛关注,废弃塑料在自然界里分解速度很慢,完全分解要几十年甚至上百年,造成环境污染。因此,全生物降解塑料越来越受到国内外诸多学者和业界人士的广泛关注。然而,目前可应用于全生物降解的基体树脂主要是PBAT(聚己二酸/对苯二甲酸丁二酯)、PLA(聚乳酸)、PHA(聚羟基脂肪酸酯)和PPC(聚碳酸亚丙酯)等,采用其制备的全生物降解塑料可广泛应用于超市购物袋、外卖餐盒、农用地膜等领域。但是,研究发现采用单独的一种基体树脂制备的全生物降解塑料不仅价格高昂,而且在力学性能、热性能、阻隔性能及生产成本等方面也难以满足产品的要求,存在着诸多问题。
苎麻具备纤维产量高、富含木质素、纤维强度大、可完全降解等特性,属于可再生资源,因此具备作为全生物降解材料的的潜力。但是由于苎麻中的纤维素、半纤维素和木质素表面含有大量的氢键,具有较强的极性,而PBAT极性较弱,若将苎麻和PBAT联用会出现纤维素与PBAT树脂在混合过程中不能很好的相容的问题,容易出现分层现象,得到的产品晶点也较多,分布不均,而且还会影响产品的性能,导致产品的机械性能下降。因此,如何将苎麻与PBAT联用制备一种全生物降解塑料是本领域技术人员亟待解决的技术问题。
发明内容
为解决现有技术中的上述问题,本发明提供了一种全生物降解的苎麻改性材料及其制备方法。
为实现上述目的,本发明提供了如下技术方案:
本发明的技术方案之一:
本发明提供了一种全生物降解的苎麻改性材料,所述苎麻改性材料包括以下原料:苎麻粉、聚己二酸/对苯二甲酸丁二酯(PBAT)、聚乳酸(PLA)、偶联剂、无水乙醇和环戊油。
有益效果:本发明在制备全生物降解塑料时加入环戊油,其可在苎麻改性材料制备过程中受热挥发产生气泡,形成多孔结构,降低材料密度的同时保持机械强度;而且其形成的多孔结构在后续的降解过程中能够增加微生物的接触面积,增强氧气渗透性,进一步提高降解速率。
无水乙醇作为极性溶剂,可有效溶解生物降解塑料的原料或中间体,促进解聚反应均匀进行。
进一步地,所述聚己二酸/对苯二甲酸丁二酯和聚乳酸的添加量为原料总质量的70wt.%-87wt.%;
所述苎麻粉的添加量为原料总质量的10wt.%-30wt.%;
所述偶联剂的添加量为原料总质量的0.5wt.%-0.8wt.%;
所述无水乙醇的添加量为原料总质量的2.0wt.%-3.0wt.%;本申请中无水乙醇(≥99.9%)的添加量需精确控制在2.0wt.%-3.0wt.%,过量会延缓降解速率,降低产品的机械强度。
所述环戊油的添加量为原料总质量的0.5wt.%-2.0wt.%,过量会导致产品的强度下降或降解过快。
更进一步地,所述聚己二酸/对苯二甲酸丁二酯和聚乳酸的质量比为0.6-0.8。
更进一步地,所述苎麻粉的粒度为500-700目。
更进一步地,所述偶联剂为硅烷偶联剂或铝酸化合物,优选为LS60、KH550或KH560。
本发明的技术方案之二:本发明还提供了一种全生物降解的苎麻改性材料的制备方法,包括以下步骤:
(1)将所述苎麻粉和偶联剂在搅拌状态下进行一次混炼,然后加入所述聚己二酸/对苯二甲酸丁二酯和聚乳酸进行二次混炼,最后加入无水乙醇和环戊油经过三次混炼后得到混合料;
(2)将所述混合料经过挤出和注射成型后得到所述苎麻改性材料。
进一步地,步骤(1)中所述搅拌的转速为30rpm;
所述一次混炼的时间为15min;
所述二次混炼的时间为5min;
所述三次混炼的时间为4min。
进一步地,步骤(1)中所述苎麻粉是在搅拌状态下与所述偶联剂混合,搅拌的转速为20rpm。
进一步地,步骤(2)中所述挤出过程设置挤出机各区温度为:170℃、170℃、170℃、175℃、175℃、175℃、175℃、175℃、175℃,转速为150rpm,主喂料频率为4.0Hz,主机电流为13A。本发明中的混合料经过挤出机的机械剪切和压缩作用,将混合料进行熔融并塑化,确保材料均匀性。
进一步地,步骤(2)中所述注射成型过程设置注射机各区温度为180℃、190℃、200℃,注射压力为125bar,射胶流量为60%。
本发明在制备全生物降解的苎麻改性材料时可以加入成核剂诱导结晶,进一步提高所述苎麻改性材料的结晶温度。
进一步地,所述成核剂的添加量为0.1%-0.3%,加入成核剂后能够使结晶温度从68℃提升至86℃。
本发明与现有技术相比的有益效果为:
本发明针对纤维分子的特有晶型结构,开展苎麻改性材料的配方设计、调整工艺参数,使苎麻中的纤维素分子能够完美与基体树脂实现两相界面的共混共熔,可实现苎麻纤维素的高质量占比的填充,使样品性能稳定,降低生产成本,提升市场竞争力。
本发明在制备苎麻改性材料过程中加入偶联剂可以活化苎麻粉体,活化后的苎麻粉体可以填充基体树脂PBAT/PLA,制备全生物降解高分子材料。而且本发明制备的苎麻改性材料的拉伸强度和弯曲强度可高达58.4MPa和110.8MPa,冲击强度可达40.6KJ/m2,可以满足不同客户群体的实际应用。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。
图1为本发明中的天然苎麻粉和实施例1、实施例9、实施例10中分别经过偶联剂LS60、KH550和KH560活化后苎麻粉的表面化学结构的红外光谱图;

图1
图2为本发明实施例12-14和对比例制备的苎麻改性材料截面的SEM图,其中,图a为对比例制备的苎麻改性材料截面的SEM图,图b为实施例12制备的苎麻改性材料截面的SEM图,图c为实施例14制备的苎麻改性材料截面的SEM图,图d为实施例13制备的苎麻改性材料截面的SEM图;

图2
图3为本发明实施例1-9制备的苎麻改性材料的维卡软化点示意图;

图3
图4为本发明实施例1-9制备的苎麻改性材料的拉伸强度柱状图;

图4
图5为本发明实施例1-9制备的苎麻改性材料的弯曲强度柱状图;

图5
图6为本发明实施例1-9制备的苎麻改性材料的冲击强度柱状图。

图6
具体实施方式
现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为还具体公开了该范围的上限和下限之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的文献冲突时,以本说明书的内容为准。
在不背离本发明的范围或精神的情况下,可对本发明说明书的具体实施方式做多种改进和变化,这对本领域技术人员而言是显而易见的。由本发明的说明书得到的其他实施方式对技术人员而言是显而易见的。本发明说明书和实施例仅是示例性的。
关于本文中所使用的“包含”、“包括”、“具有”、“含有”等等,均为开放性的用语,即意指包含但不限于。
本发明以下实施例中所使用的原料均可通过市售购得。
实施例1
一种全生物降解的苎麻改性材料的制备方法,包括以下步骤:
(1)对高速混合机进行预热,待高速混合机的温度达到120℃时,加入10wt.%的粒度为500-700目的苎麻粉,将转速调至20rpm,加入0.4wt.%的偶联剂KH560,接着再将转速调至30rpm进行混炼15min,然后加入86.8wt.%的PLA和PBAT树脂(其中,PBAT与PLA的质量比0.8)继续混炼5min,最后加入2.0wt.%的无水乙醇和0.8wt.%的环戊油混炼4min,卸料,得到混合料。
(2)打开挤出机电源,对挤出机进行预热,设置各区温度依次为170℃、170℃、170℃、175℃、175℃、175℃、175℃、175℃、175℃,主机转速为150rpm,主喂料频率为4.0Hz,主机电流为13A,将上述混合料放入挤出机中进行挤出,得到熔融物料。
(3)设置注射机的各区温度为180℃、190℃、200℃,注射压力为125bar,射胶流量为60%,将上述熔融物料通过注射机注射成型,得到苎麻改性材料的样条。
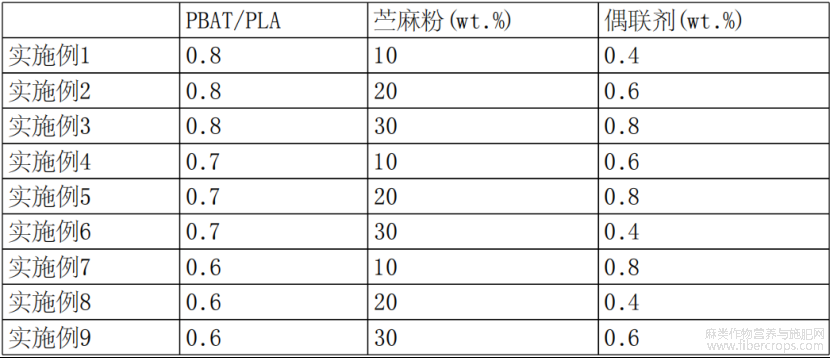
实施例2-9提供一种全生物降解的苎麻改性材料的制备方法,其制备步骤与实施例1的区别为:PBAT和PLA的质量比不同,苎麻粉和偶联剂的添加量不同,实施例2-9的PBAT和PLA的质量比、苎麻粉和偶联剂的添加量如表1所示;
其余原料和制备方法与实施例1均相同。
表1
实施例10
一种全生物降解的苎麻改性材料的制备方法,与实施例1的区别在于:偶联剂为KH550;其余步骤同实施例1。
实施例11
一种全生物降解的苎麻改性材料的制备方法,与实施例1的区别在于:偶联剂为LS60;
其余步骤同实施例1。
实施例12
一种全生物降解的苎麻改性材料的制备方法,与实施例1的区别在于:
PLA与PBAT的质量比为6∶4,苎麻粉的加入量为30wt.%,偶联剂的加入量为0.4wt.%;其余步骤同实施例1。
实施例13
一种全生物降解的苎麻改性材料的制备方法,与实施例12的区别在于:偶联剂的加入量为0.6wt.%;
其余步骤同实施例12。
实施例14
一种全生物降解的苎麻改性材料的制备方法,与实施例12的区别在于:偶联剂的加入量为0.8wt.%;
其余步骤同实施例12。
对比例
一种全生物降解的苎麻改性材料的制备方法,与实施例12的区别在于:偶联剂的加入量为0;
其余步骤同实施例12。
性能测试:
1、红外光谱测试
用破碎机将实施例1、实施例10和实施例11制备的苎麻改性材料粉碎成100目左右的细粉,按质量比为1∶100的比例分别称取样品粉末和KBr,在玛瑙研钵内研磨均匀,同时按质量比为1∶100的比例称取天然苎麻粉和KBr,在玛瑙研钵内研磨均匀。采用溴化钾压片法,使用傅里叶变换红外光谱仪(Fourier transform infrared spectrometer,FTIR)(Nicolet Impact 420,美国)对压片后的样品进行测试,采用FTIR测试的天然苎麻粉和分别经LS60、KH550和KH560活化后苎麻粉的表面化学结构的红外光谱图如图1所示。
由图1可以看出,与未处理的天然苎麻粉相比,经LS60活化后的苎麻粉在2850cm-1处明显多了一个吸收峰,这是亚甲基的伸缩振动,说明LS60成功接枝到了苎麻粉上,植物纤维中的酯、有机酸类等物质被碱溶液去除;在波数为3400cm-1位置为-OH的吸收峰,苎麻纤维表面羟基较多,所以红外光谱图此处的峰非常宽。但是经过KH550和KH560活化处理后的苎麻粉体在此波数处的吸收峰明显变窄,说明苎麻纤维的极性降低,此外,图1显示在波数为1030cm-1处有明显的振动峰,此处为Si-O的伸缩振动峰,说明偶联剂KH550和KH560可以很好地将苎麻粉体包覆起来。
2、微观形貌
将实施例12-14和对比例制备的苎麻改性材料在液氮中进行脆断,对苎麻改性材料的表面、断面进行喷金处理后,使用场发射扫描电子显微镜(field emission scanning electron microscope,FESEM)(Gemini SEM 300,Zeiss,德国)观察样品的微观形貌,观察粉体在基体树脂中的分散状况,加速电压为3.0kV。
本发明实施例12-14和对比例制备的苎麻改性材料截面的SEM图如图2所示,可以发现未添加偶联剂的苎麻改性材料(如图a所示)中,苎麻粉体在基体树脂中出现了大量团聚,甚至可以看到很多粗纤维,并且基体树脂中存在明显的空洞,出现了相分离现象。当偶联剂KH560的加入量为0.4wt.%(如图b所示),苎麻纤维由于被KH560活化,其极性降低,能够较好地分布在基体树脂中。当偶联剂KH560的加入量为0.8wt.%(如图c所示),苎麻粉体表面包覆了过量的KH560,KH560亲水基团因无法直接参与和苎麻的反应而裸露在外面,致使样品的截面又出现了部分断层现象。当偶联剂KH560的加入量为0.6wt.%(如图d所示),苎麻粉体表面的-OH与KH560的硅醇键充分的结合,粉体基本处于弱亲水特性,与基体树脂相容性很好,未发现有苎麻粉体团聚和出现在基体树脂表面。
3、维卡软化点测试
采用热变形维卡温度测定仪(XRW-300C-3,承德金博电子有限公司)分析实施例1-9制备的苎麻改性材料的热稳定性。热变形测试按ASTM D64测试,负荷为0.45MPa,样品选择平放,尺寸为64mm×10mm×4mm,升温速率为120℃/h,每组样品测量3次,取平均值。
实施例1-9制备的苎麻改性材料的维卡软化点示意图如图3所示,由图3可以知晓,当苎麻粉添加量为10wt.%时,材料的维卡软化点相对较高,因为苎麻纤维结构蓬松,内部空洞较多,在一定压力条件下,很容易引起其变形。当基体树脂中PBAT含量较高时,材料的维卡软化点相对较低,因为PBAT自身的熔点仅有107℃左右,远低于PLA的熔点,更容易变形;当PBAT/PLA为0.7,苎麻粉体填充量为10wt.%,偶联剂含量为0.6wt.%时,材料的热稳定较好,热变形温度可达83.4℃。本申请还可以为了提高结晶效率,在改性材料中添加少量的成核剂诱导结晶,进一步提高改性材料的结晶温度。
4、熔融指数
熔融指数(Melt Flow Rate,MFR)是一种表示高分子材料加工时的流动性的数值。是在一定的条件下,单位时间流过某一孔洞的塑料质量。熔融指数的值越大,表示该塑胶材料的加工流动性越好,反之则越差。
将实施例1-9制备的苎麻改性材料在190℃,2.16kg压力条件下,通过直径为2.1mm的圆管流出,计算10min内熔体流出的质量,结果如表2所示。
表2
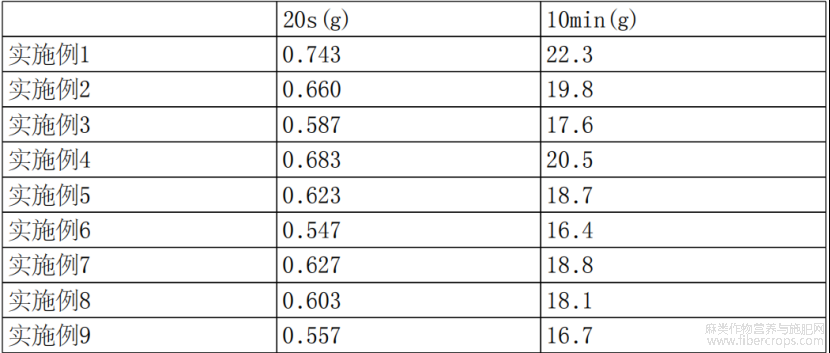
纯PLA树脂的熔融指数约为30g/10min,纯PBAT树脂的熔融指数为4.5g/10min。从表2可以知晓,在基体树脂中当PBAT/PLA的质量占比由0.8下降到0.6,苎麻改性材料的熔融指数也相应出现下降趋势。当苎麻粉体的整体占比从10wt.%提高到30wt.%,苎麻改性材料的流动性变差,主要原因是苎麻粉体无规的排列在基体树脂中阻碍了基体树脂的流动。偶联剂的占比提高,一方面可以充分包覆粉体颗粒,使苎麻粉体与基体树脂具有很好的相容性,同时偶联剂还起到润滑作用,有利于苎麻改性材料流动性的提升。
5、机械性能
力学性能是植物纤维/聚合物复合材料的一项主要技术指标,它在很大程度上决定了此类复合材料的使用寿命。因而,大量文献报道了此类复合材料的力学性能研究。
本发明根据GB/T 1040-2018对实施例1-9制备的苎麻改性材料的拉伸性能进行测试,测试速度为50mm/min;根据GB/T 9431-2008对实施例1-9制备的苎麻改性材料的弯曲性能进行测试,测试速度为50mm/min;根据GB/T 1843-2008对实施例1-9制备的苎麻改性材料的冲击性能进行测试。其中,采用拉力试验机对苎麻改性材料进行拉伸强度和弯曲强度测试,缺口冲击强度采用悬臂梁冲击试验机按GB/T 1043—2008进行测试。每组样品测试5次,结果取平均值。其中,拉伸强度如图4所示,弯曲强度如图5所示,冲击强度如图6所示。
通过图4-6可以知晓本发明制备的改性苎麻材料在基体树脂、苎麻粉和改性剂的共同作用下,其拉伸轻度和弯曲强度得到显著提高,拉伸强度可高达58.4MPa,弯曲强度可高达110.8MPa;在冲击强度方面,因为PBAT具有一定的韧性,所以材料的冲击强度也表现的更加优异,冲击强度可高达40.6KJ/m2,远超标准限值,可满足不同客户群体的实际需求。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
文章摘自国家发明专利,一种全生物降解的苎麻改性材料及其制备方法,发明人:汪红武,郑华明,张奥深,陈炼,柳威,王宇航,熊伟,申请号:202511571138.1,申请日:2025.10.30。
