摘 要:本发明属于检测技术领域,尤其涉及一种亚麻子药材、饮片、提取物及其制剂HPLC特征图谱的构建方法及其应用。本发明提供的HPLC特征图谱构建方法包括以下步骤:准备供试品,所述供试品为亚麻子药材、亚麻子饮片、提取物及其制剂;将所述供试品溶解,得到供试品溶液;将所述供试品溶液采用高效液相色谱法测定,得到对应供试品的HPLC特征图谱;所述高效液相色谱法的色谱条件为:色谱柱为C18柱;流动相A为乙腈,流动相B为0.2vol%乙酸溶液,梯度洗脱。本发明提供的特征图谱构建方法可以为亚麻子药材、饮片、提取物及其制剂的鉴别提供更为科学的依据。
技术要点
1.一种亚麻子药材、饮片、提取物、制剂HPLC特征图谱的构建方法,其特征在于,包括以下步骤:
准备供试品,所述供试品为亚麻子药材、亚麻子饮片、亚麻子提取物及亚麻子制剂;
将所述供试品溶解,得到供试品溶液;
将所述供试品溶液采用高效液相色谱法测定,得到对应供试品的HPLC特征图谱;
所述高效液相色谱法的色谱条件为:色谱柱为C18柱;流动相A为乙腈,流动相B为0.2vol%乙酸溶液,梯度洗脱。
2.根据权利要求1所述的构建方法,其特征在于,所述梯度洗脱具体为:
0~7min,A相:1vol%,B相:99vol%;
7~20min,A相:1~4vol%,B相:99~96vol%;
20~28min,A相:4vol%,B相:96vol%;
28~36min,A相:4~10vol%,B相:96~90vol%;
36~48min,A相:10~20vol%,B相:90~80vol%。
3.根据权利要求1所述的构建方法,其特征在于,所述溶解的溶剂为30vol%的甲醇溶液;所述溶解在超声辅助下进行,所述超声辅助的功率为580~620W,频率为35~45kHz,时间为25~35min。
4.根据权利要求1所述的构建方法,其特征在于,所述高效液相色谱法的色谱条件还包括:流动相的流速为0.8mL/min;检测波长为278nm;进样量为10μL;柱温30℃;理论板数按色氨酸峰计算不低于3000。
5.根据权利要求1所述的构建方法,其特征在于,还包括以下步骤:
将亚麻子对照药材溶解,得到对照药材参照物溶液;将色氨酸溶解,得到对照品参照物溶液;
将所述对照药材参照物溶液和对照品参照物溶液采用高效液相色谱法测定,得到参照物的色谱图;并根据所述参照物的色谱图对供试品的HPLC特征图谱的成分进行定性。
6.根据权利要求5所述的构建方法,其特征在于,还包括以下步骤:
采用中药色谱指纹图谱相似度评价系统对亚麻子药材的HPLC特征图谱的相似度进行评价,得到由6个特征峰构成的亚麻子药材的HPLC标准特征图谱;在所述HPLC标准特征图谱中,与色氨酸对照品参照物峰相对应的峰6为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
7.根据权利要求5所述的构建方法,其特征在于,还包括以下步骤:
采用中药色谱指纹图谱相似度评价系统对亚麻子饮片的HPLC特征图谱的相似度进行评价,得到由6个特征峰构成的亚麻子饮片的HPLC标准特征图谱;在所述HPLC标准特征图谱中,与色氨酸对照品参照物峰相对应的峰6为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
8.根据权利要求5所述的构建方法,其特征在于,还包括以下步骤:
采用中药色谱指纹图谱相似度评价系统对亚麻子提取物的HPLC特征图谱的相似度进行评价,得到由6个特征峰构成的亚麻子提取物的HPLC标准特征图谱;在所述HPLC标准特征图谱中,与色氨酸对照品参照物峰相对应的峰6为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
9.根据权利要求5所述的构建方法,其特征在于,还包括以下步骤:
采用中药色谱指纹图谱相似度评价系统对亚麻子制剂的HPLC特征图谱的相似度进行评价,得到由6个特征峰构成的亚麻子制剂的HPLC标准特征图谱;在所述HPLC标准特征图谱中,与色氨酸对照品参照物峰相对应的峰6为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
10.一种亚麻子药材、饮片、提取物、制剂的鉴别方法,其特征在于,采用权利要求1~9任一项所述的构建方法获得的HPLC特征图谱作为鉴别亚麻子药材、饮片、提取物及制剂的依据。
技术领域
本发明属于检测技术领域,尤其涉及一种亚麻子药材、饮片、提取物、及其制剂HPLC特征图谱的构建方法及其应用。
背景技术
亚麻子为亚麻科植物亚麻Linum usitatissimum L.的干燥成熟种子,具有润燥,祛风之功效。为了确保亚麻子药材、饮片、提取物及配方颗粒其质量的均一、稳定,建立一种新的特征图谱方法对其质量进行把控,是十分必要的。
发明内容
有鉴于此,本发明的目的在于提供一种亚麻子药材、饮片、提取物及其制剂HPLC特征图谱的构建方法及其应用,本发明提供的构建方法可为亚麻子药材、饮片、提取物及其制剂的鉴别提供更为科学的依据。
本发明提供了一种亚麻子药材、饮片、提取物、制剂HPLC特征图谱的构建方法,包括以下步骤:
准备供试品,所述供试品为亚麻子药材、亚麻子饮片、亚麻子提取物及其制剂;
将所述供试品溶解,得到供试品溶液;
将所述供试品溶液采用高效液相色谱法测定,得到对应供试品的HPLC特征图谱;
所述高效液相色谱法的色谱条件为:色谱柱为C18柱;流动相A为乙腈,流动相B为0.2vol%乙酸溶液,梯度洗脱。
在本发明提供的构建方法中,所述亚麻子饮片是由亚麻子药材经炮制,即除去杂质得到的亚麻子饮片;所述亚麻子提取物是由亚麻子饮片提取制备而成的冻干粉;所述亚麻子制剂是由亚麻子提取物经浓缩制成的颗粒。
在本发明提供的构建方法中,所述溶解的溶剂优选为30vol%的甲醇溶液,该溶剂所得的色谱峰信息量大,分离度好。
在本发明提供的构建方法中,所述溶解优选在超声辅助下进行;所述超声辅助的功率优选为580~620W,更优选为600W;所述超声辅助的频率优选为35~45kHz,更优选为40kHz;所述超声辅助的时间优选为25~35min,更优选为30min。
在本发明提供的构建方法中,当所述供试品为亚麻子药材或亚麻子饮片时,溶解制备供试品溶液的具体过程优选包括:将供试品加水煎煮,滤过,滤液蒸干,蒸干残渣与溶剂混合,超声辅助溶解,放冷,摇匀,滤过,得到的续滤液即为供试品溶液;其中,所述供试品与水的用量比优选为(0.8~1.2)g:50mL,更优选为1g:50mL;所述煎煮的时间优选为25~35min,更优选为28~32min,再优选为30min;所述溶剂与供试品的用量比优选为25mL:(0.8~1.2)g,更优选为25mL:1g。
在本发明提供的构建方法中,当所述供试品为亚麻子提取物时,溶解制备供试品溶液的具体过程优选包括:将供试品与溶剂混合,超声辅助溶解,放冷,摇匀,滤过,得到的续滤液即为供试品溶液;其中,所述供试品与溶剂的用量比优选为(0.3~0.7)g:25mL,更优选0.5g:25mL。
在本发明提供的构建方法中,当所述供试品为亚麻子制剂时,溶解制备供试品溶液的具体过程优选包括:将供试品研细后与溶剂混合,超声辅助溶解,放冷,摇匀,滤过,得到的续滤液即为供试品溶液;其中,所述供试品与溶剂的用量比优选为(0.3~0.7)g:25mL,更优选0.5g:25mL。
在本发明提供的构建方法中,进行所述高效液相色谱法测定时,所述梯度洗脱具体过程优选为:
0~7min,A相:1vol%,B相:99vol%;
7~20min,A相:1~4vol%,B相:99~96vol%;
20~28min,A相:4vol%,B相:96vol%;
28~36min,A相:4~10vol%,B相:96~90vol%;
36~48min,A相:10~20vol%,B相:90~80vol%。
在本发明提供的构建方法中,进行所述高效液相色谱法测定时,流动相的流速优选为0.8mL/min,在此流速条件下获得的色谱图峰形较好,基线更平稳。
在本发明提供的构建方法中,进行所述高效液相色谱法测定时,检测波长优选为278nm,在此检测波长条件下获得的色谱图信息量较大,基线更平稳。
在本发明提供的构建方法中,进行所述高效液相色谱法测定时,进样量优选为10μL。
在本发明提供的构建方法中,进行所述高效液相色谱法测定时,色谱柱中的填充剂为十八烷基硅烷键合硅胶;所述色谱柱的柱长优选为200~300mm,更优选为为250mm;所述色谱柱的内径优选为4~5mm,更优选为4.6mm;所述色谱柱的填充剂粒径优选为3~7μm,更优选为5μm。
在本发明提供的构建方法中,进行所述高效液相色谱法测定时,所述色谱柱的柱温优选为30℃,在此柱温条件下获得的色谱图的峰形较为对称且保留时间较为适宜。
在本发明提供的构建方法中,进行所述高效液相色谱法测定时,理论板数按色氨酸峰计算应不低于3000。
在本发明提供的构建方法中,优选还包括以下步骤:
将亚麻子对照药材溶解,得到对照药材参照物溶液;将色氨酸溶解,得到对照品参照物溶液;
将所述对照药材参照物溶液和对照品参照物溶液采用高效液相色谱法测定,得到参照物的色谱图;并根据所述参照物的色谱图对供试品的HPLC特征图谱的成分进行定性。
在本发明提供的构建方法中,溶解制备所述对照药材参照物溶液的具体过程优选包括:将亚麻子对照药材加水煎煮,滤过,滤液蒸干,蒸干残渣与溶剂混合,超声辅助溶解,放冷,摇匀,滤过,得到的续滤液即为对照药材参照物溶液;其中,所述亚麻子对照药材与水的用量比优选为(0.8~1.2)g:50mL,更优选为1g:50mL;所述煎煮的时间优选为25~35min,更优选为28~32min,再优选为30min;所述溶剂优选为30vol%的甲醇溶液;所述溶剂与亚麻子对照药材的用量比优选为25mL:(0.8~1.2)g,更优选为25mL:1g;所述超声辅助的功率优选为580~620W,更优选为600W;所述超声辅助的频率优选为35~45kHz,更优选为40kHz;所述超声辅助的时间优选为25~35min,更优选为30min。
在本发明提供的构建方法中,溶解制备所述对照品参照物溶液的具体过程优选包括:将色氨酸与溶剂混合,得到对照品参照物溶液;其中,所述溶剂优选为30vol%的甲醇溶液;所述色氨酸与溶剂的用量比优选为(8~12)μg:1mL,更优选为10μg:1mL。
在本发明提供的构建方法中,优选还包括以下步骤:
采用中药色谱指纹图谱相似度评价系统对亚麻子药材的HPLC特征图谱的相似度进行评价,得到由6个特征峰构成的亚麻子药材的HPLC标准特征图谱;在所述HPLC标准特征图谱中,与色氨酸对照品参照物峰相对应的峰6为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
在本发明提供的构建方法中,优选还包括以下步骤:
采用中药色谱指纹图谱相似度评价系统对亚麻子饮片的HPLC特征图谱的相似度进行评价,得到由6个特征峰构成的亚麻子饮片的HPLC标准特征图谱;在所述HPLC标准特征图谱中,与色氨酸对照品参照物峰相对应的峰6为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
在本发明提供的构建方法中,优选还包括以下步骤:
采用中药色谱指纹图谱相似度评价系统对亚麻子提取物的HPLC特征图谱的相似度进行评价,得到由6个特征峰构成的亚麻子提取物的HPLC标准特征图谱;在所述HPLC标准特征图谱中,与色氨酸对照品参照物峰相对应的峰6为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
在本发明提供的构建方法中,优选还包括以下步骤:
采用中药色谱指纹图谱相似度评价系统对亚麻子制剂的HPLC特征图谱的相似度进行评价,得到由6个特征峰构成的亚麻子制剂的HPLC标准特征图谱;在所述HPLC标准特征图谱中,与色氨酸对照品参照物峰相对应的峰6为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)
本发明还提供了一种亚麻子药材、饮片、提取物、制剂的鉴别方法,该方法采用上述技术方案所述的构建方法获得的HPLC特征图谱作为鉴别亚麻子药材、饮片、提取物、制剂的依据。
与现有技术相比,本发明提供了一种亚麻子药材、饮片、提取物、制剂HPLC特征图谱的构建方法及其应用,本发明提供的方法能够用于建立亚麻子药材、饮片、提取物以及制剂的HPLC特征图谱,具有良好的重复性、精密度和稳定性,可以为亚麻子药材、饮片、提取物及制剂的鉴别提供更为科学的依据。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
图1是本发明实施例1提供的亚麻子提取物特征图谱色谱峰指认图;
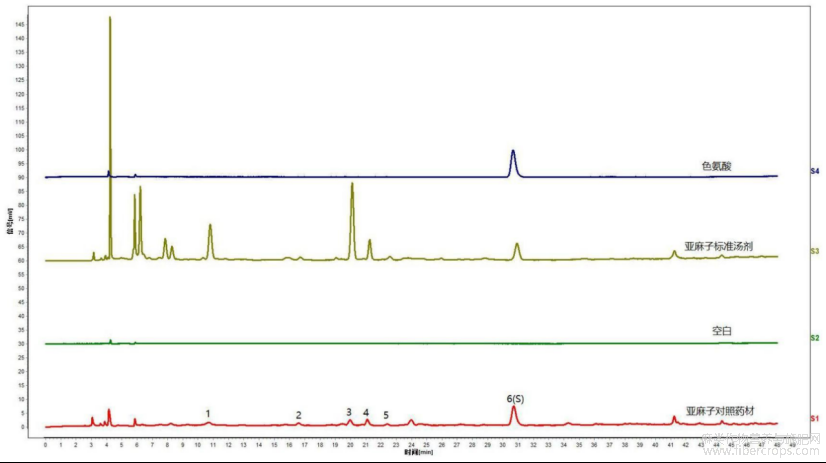
图1
图2是本发明实施例1提供的色氨酸对照品的光谱图;
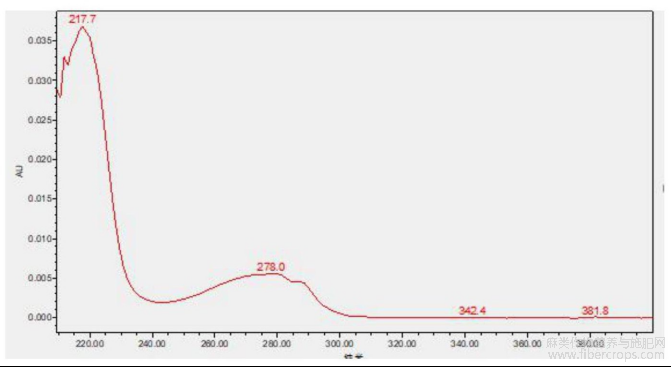
图2
图3是本发明实施例1提供的供试品中色氨酸的光谱图;
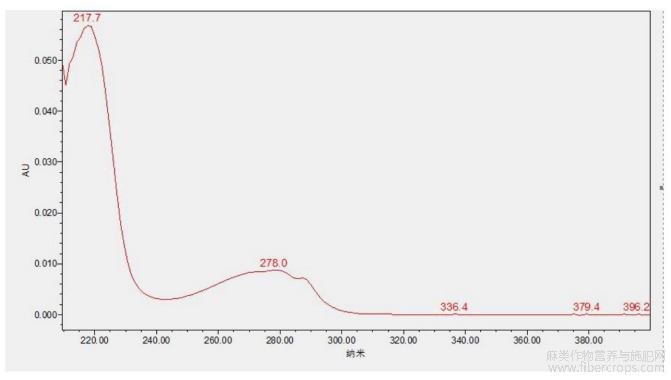
图3
图4是本发明实施例1提供的亚麻子提取物不同仪器考察的实验结果图;
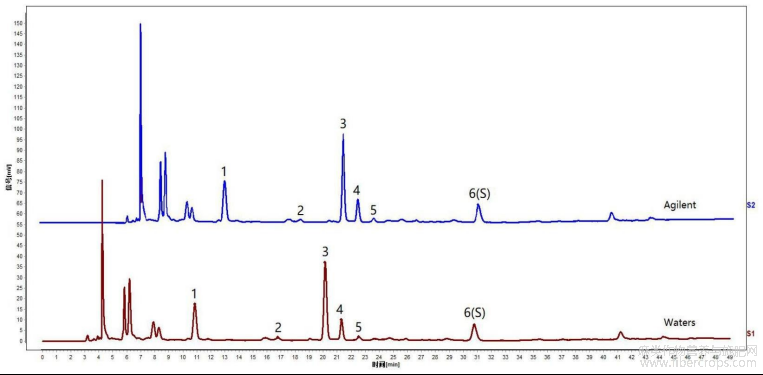
图4
图5是本发明实施例1提供的亚麻子提取物不同色谱柱考察的实验结果图;
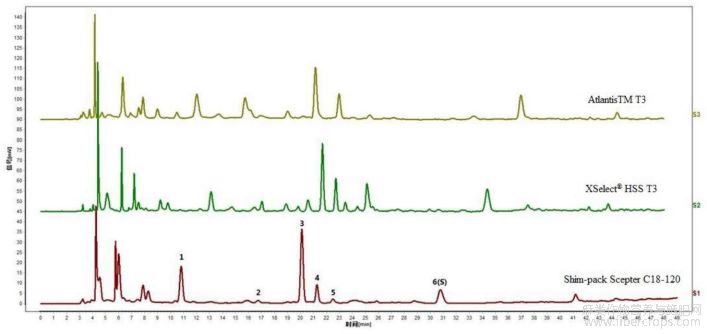
图5
图6是本发明实施例1提供的16批亚麻子提取物样品的特征图谱验证图;
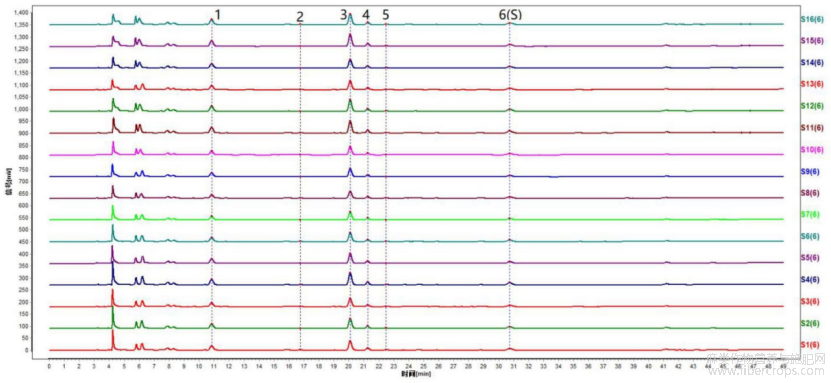
图6
图7是本发明实施例1提供的亚麻子提取物特征图谱的对照图谱;
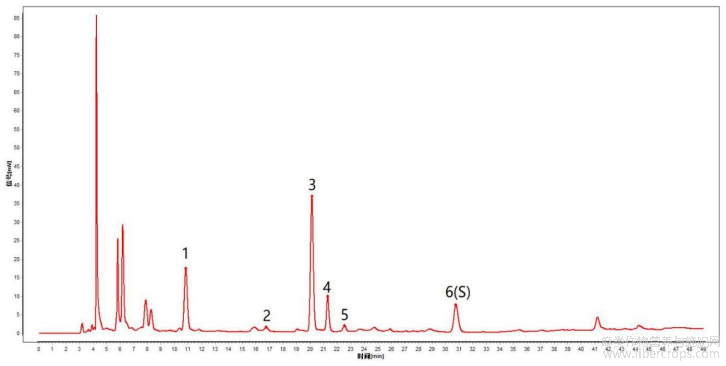
图7
图8是本发明实施例2提供的亚麻子药材供试品溶液制备过程中溶解方式考察的实验结果图;
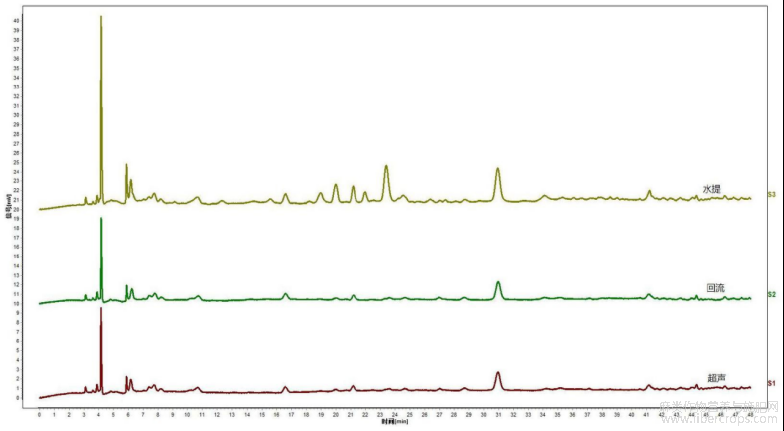
图8
图9是本发明实施例2提供的亚麻子药材特征图谱色谱峰指认图;
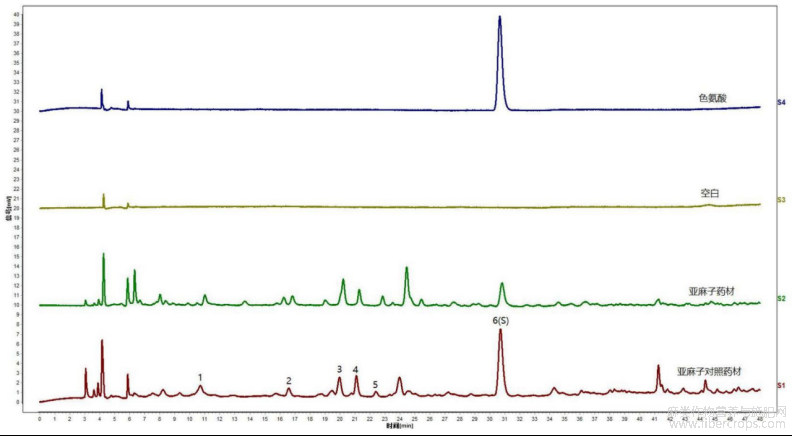
图9
图10是本发明实施例2提供的亚麻子药材不同仪器考察的实验结果图;
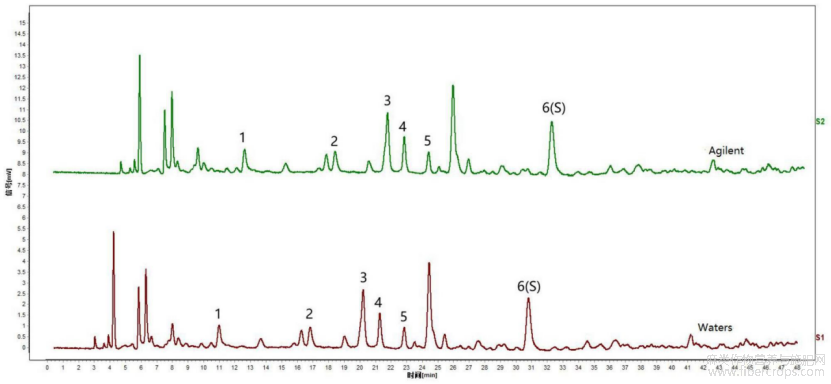
图10
图11是本发明实施例2提供的亚麻子药材不同色谱柱考察的实验结果图;
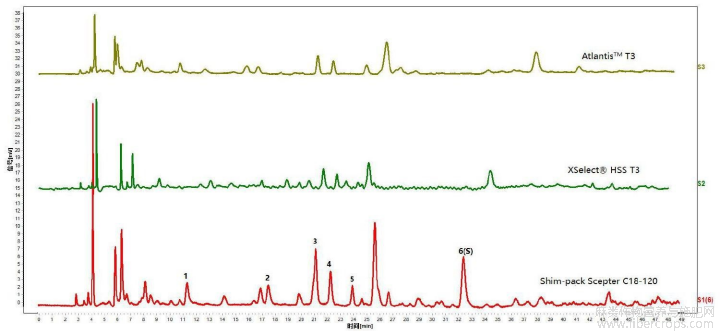
图11
图12是本发明实施例2提供的16批亚麻子药材样品的特征图谱验证图;
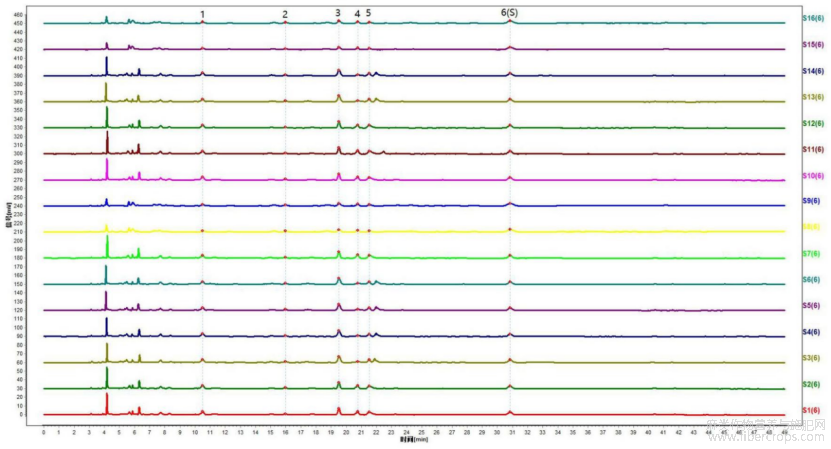
图12
图13是本发明实施例2提供的亚麻子药材特征图谱的对照图谱;
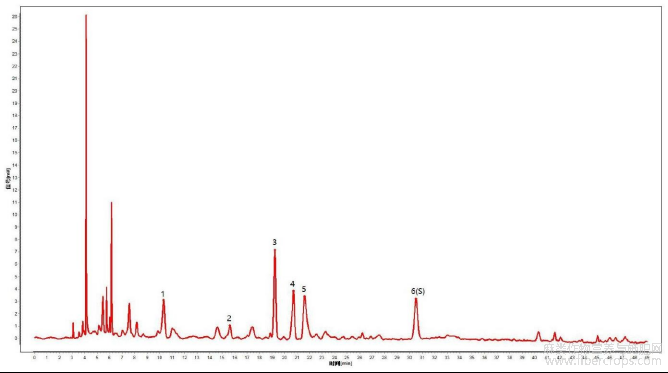
图13
图14是本发明实施例2提供的16批亚麻子饮片样品的特征图谱验证图;
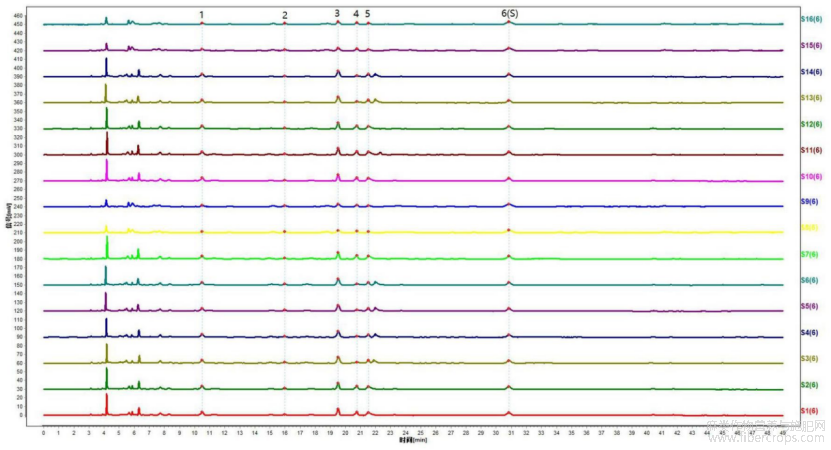
图14
图15是本发明实施例2提供的亚麻子饮片特征图谱的对照图谱;
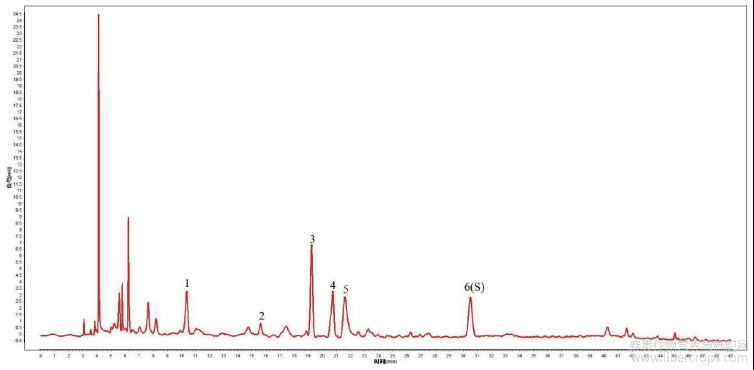
图15
图16是本发明实施例3提供的亚麻子配方颗粒不同波长色谱图;
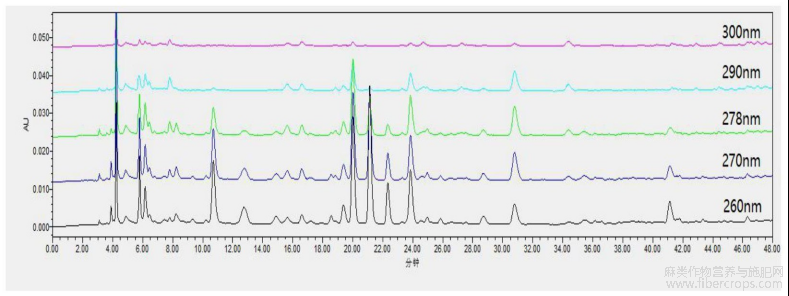
图16
图17是本发明实施例3提供的柱温考察色谱图;
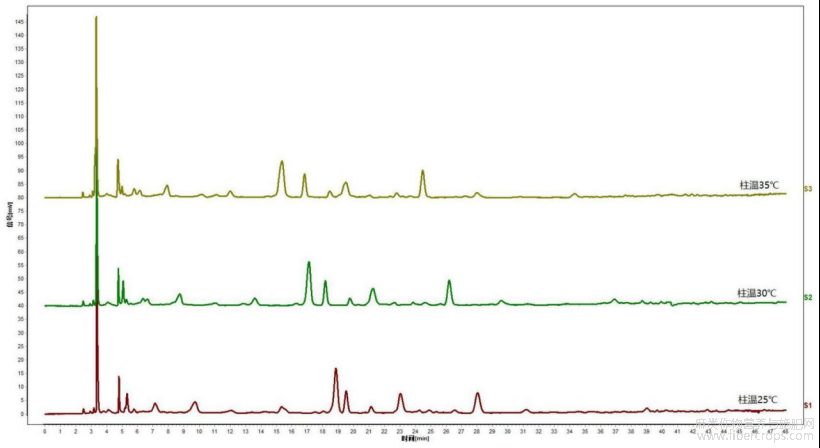
图17
图18是本发明实施例3提供的流速考察色谱图;
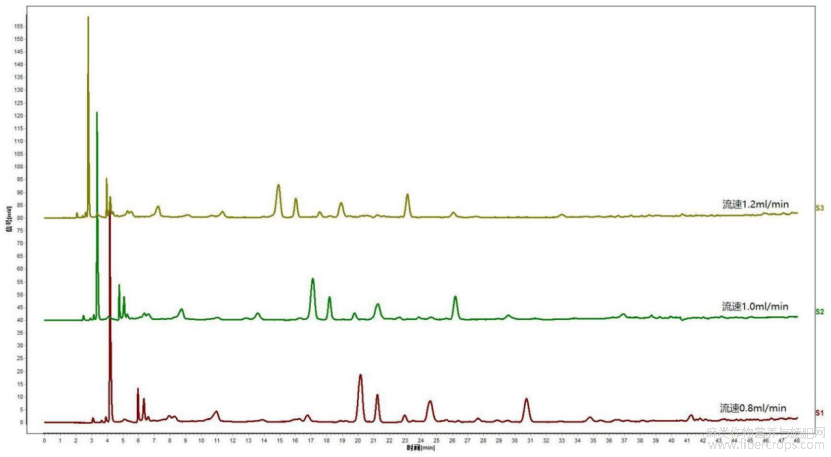
图18
图19是本发明实施例3提供的供试品溶液制备过程中溶解方式考察的实验结果图;
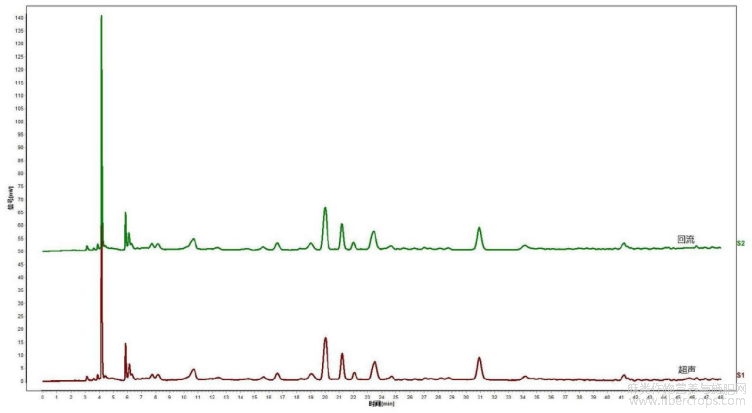
图19
图20是本发明实施例3提供的供试品溶液制备过程中溶解溶剂考察的实验结果图;
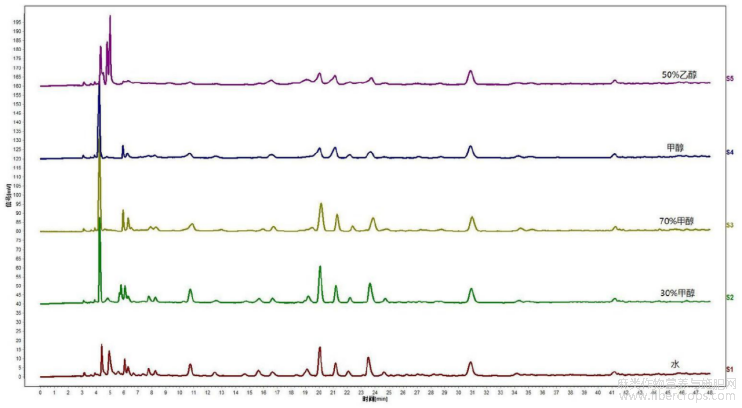
图20
图21是本发明实施例3提供的供试品溶液制备过程中溶解时间考察的实验结果图;
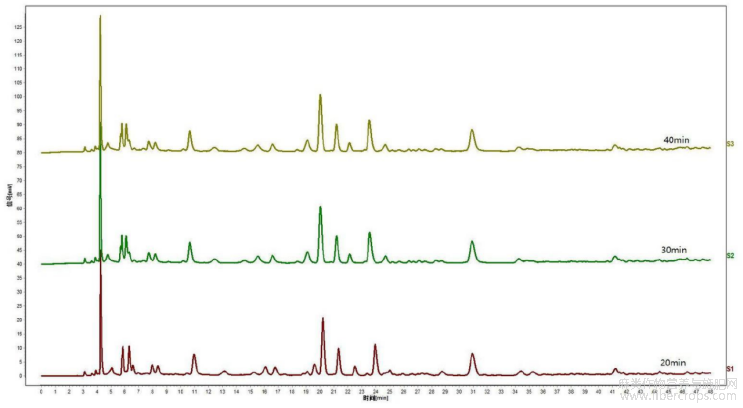
图21
图22是本发明实施例3提供的供试品溶液制备过程中溶剂加入量考察的实验结果图;
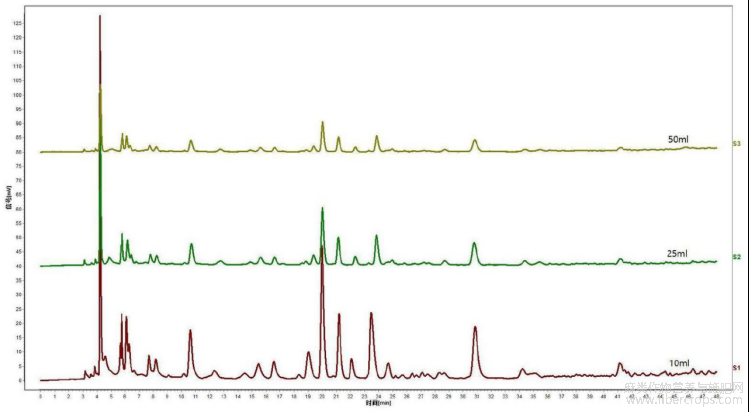
图22
图23是本发明实施例3提供的亚亚麻子配方颗粒特征图谱色谱峰指认图;
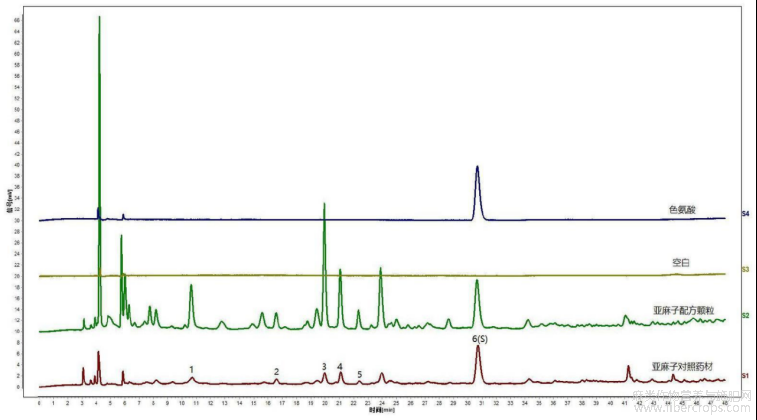
图23
图24是本发明实施例3提供的亚麻子配方颗粒不同仪器考察的实验结果图;
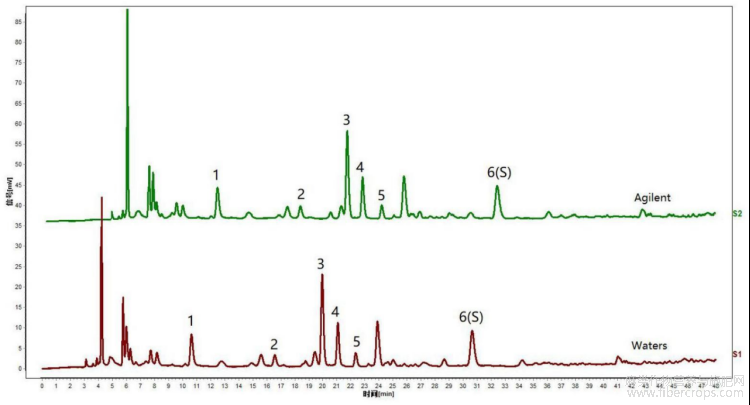
图24
图25是本发明实施例3提供的亚麻子配方颗粒不同色谱柱考察的实验结果图;
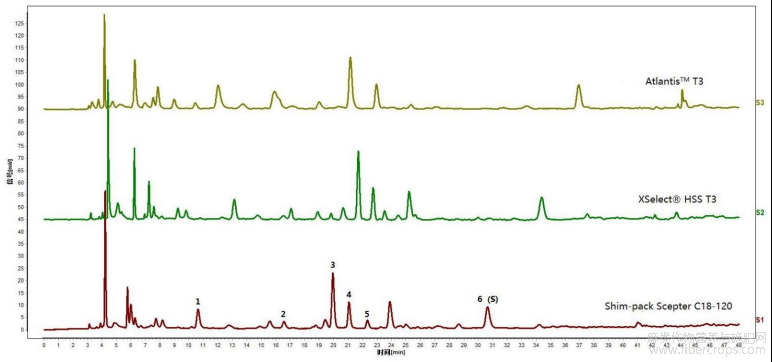
图25
图26是本发明实施例3提供的3批亚麻子配方颗粒特征图谱验证图;
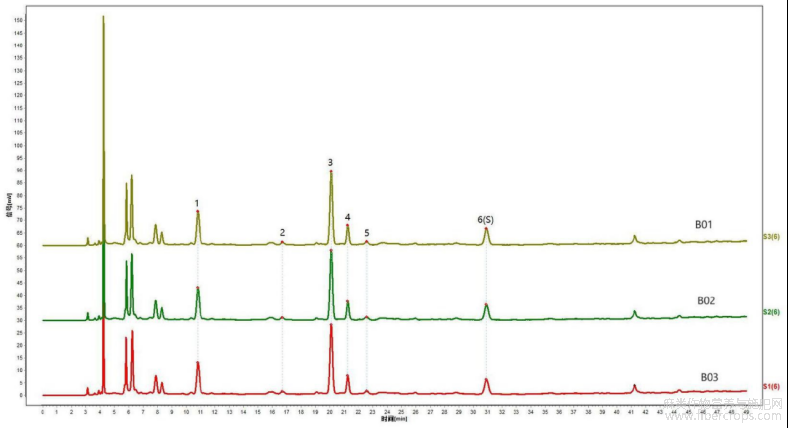
图26
图27是本发明实施例3提供的亚麻子配方颗粒特征图谱的对照图谱。
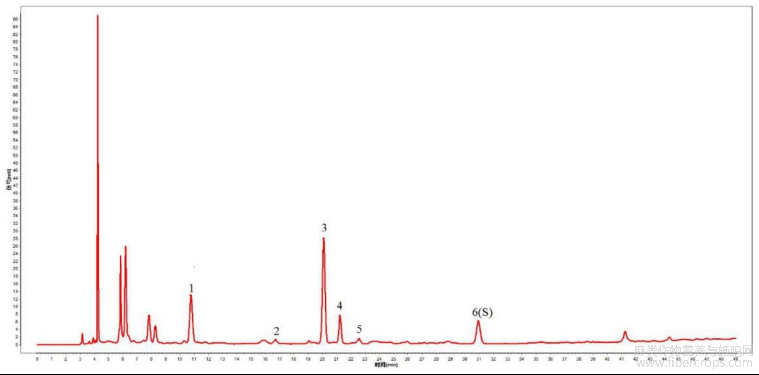
图27
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
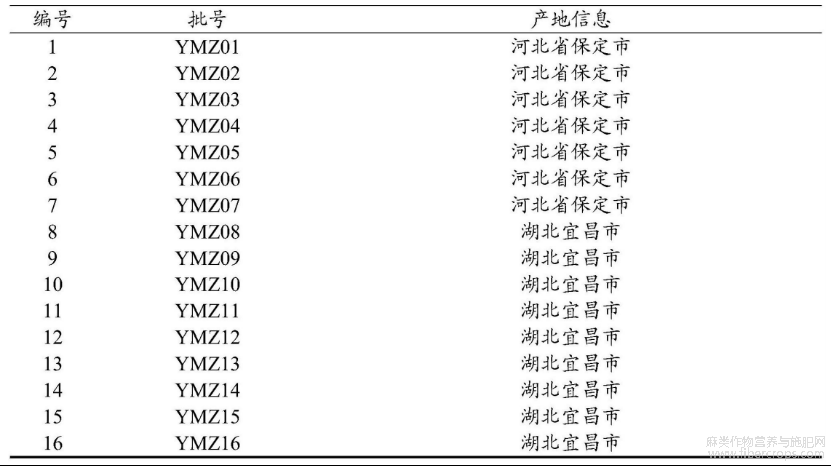
表1亚麻子药材产地信息汇总表
亚麻子饮片制备
取亚麻子药材,除去杂质,即得。将16批亚麻子药材进行炮制16批亚麻子饮片,具体对应信息见表2。
表2 16批亚麻子炮制对应表

亚麻子提取物的制备
取亚麻子饮片100g,加水煎煮两次,一煎加10倍水,浸泡30分钟,煮沸,保持微沸煎煮30分钟,200目筛网过滤,立即冷却至室温;二煎加8倍水,煮沸,保持微沸煎煮20分钟,200目筛网过滤,合并水煎液,立即冷却至室温,浓缩,冷冻干燥,分装,即得。
由16批亚麻子饮片制得16批亚麻子提取物,对应信息见下表3。
表3亚麻子提取物制备对应表

实施例1
亚麻子提取物HPLC特征图谱的构建:
实验仪器及材料
仪器:Waters、Agilent高效液相色谱仪,如无特别说明,默认使用Waters高效液相色谱仪;
电子天平:ME204E/02、MS205DU、XP26(梅特勒?托利多仪器有限公司);
超纯水机:细胞型1810A(上海摩勒科学仪器有限公司);
超声波清洗器:KQ?600DB型(600W,40KHz;昆山市超声仪器有限公司);
色谱柱:AtlantisTM T34.6×250mm(填充剂粒径5μm)、 HSS T34.6×250mm(填充剂粒径5μm)、Shim?pack Scepter C18?1204.6×250mm(填充剂粒径5μm),如无特别说明,默认使用AtlantisTM T34.6×250mm(填充剂粒径5μm)色谱柱。
2)试剂及试药
乙酸、乙腈为色谱纯,水为超纯水,色氨酸(中国食品药品检定研究院,批号:140686?202205,含量以100.0%计),其余试剂均为分析纯;
亚麻子对照药材(成都普思生物科技股份有限公司,批号:PS030308);
亚麻子提取物冻干粉(四川新绿色药业科技发展有限公司制备,批号:YMZ?BT?230601、YMZ?BT?230701、YMZ?BT?230702、YMZ?BT?230703、YMZ?BT?230704、YMZ?BT?230705、YMZ?BT?230706、YMZ?BT?230707、YMZ?BT?230708、YMZ?BT?230709、YMZ?BT?230710、YMZ?BT?230711、YMZ?BT?230712、YMZ?BT?230713、YMZ?BT?230714、YMZ?BT?230715、YMZ?BT230716)。
3)特征图谱检测方法
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A,以0.2vol%乙酸溶液为流动相B,按表4中的规定进行梯度洗脱;流速每分钟0.8mL;柱温为30℃;检测波长为278nm;理论板数按色氨酸峰计算应不低于3000。
表4梯度洗脱程序

参照物溶液的制备:取亚麻子对照药材1g,置具塞锥形瓶中,加水50mL,煎煮30分钟,滤过,滤液蒸干,残渣加入30vol%甲醇25mL,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液;另取色氨酸对照品适量,精密称定,加30vol%甲醇制成每1mL各含10μg色氨酸的溶液,作为对照品参照物溶液。
供试品溶液的制备:取本品适量,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入30vol%甲醇25mL,密塞,称定重量,超声处理(功率600W,频率40kHz)30分钟,放冷,再称定重量,用30vol%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法:分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得。
4)方法学考察
4.1)色谱峰指认
按以上拟定的实验条件,制备亚麻子提取物供试品溶液、色氨酸对照品参照物溶液、亚麻子对照药材参照物溶液,同时,参照以上拟定的实验条件,制备缺亚麻子提取物的阴性对照溶液(即,空白溶液),进行色谱检测。
对亚麻子提取物特征图谱峰进行定位,结果见图1~3,图1是本发明实施例1提供的亚麻子提取物特征图谱色谱峰指认图,图2是本发明实施例1提供的色氨酸对照品的光谱图,图3是本发明实施例1提供的供试品中色氨酸的光谱图。通过图1~3可知,色氨酸对照品的保留时间和光谱图都能和亚麻子提取物中目标峰的保留时间和光谱图一一对应,且阴性溶液无干扰,该方法专属性良好。
4.2)重复性考察
精密称取亚麻子提取物(批号:YMZ?BT?230601)6份,按拟定实验方法进行制备及测定,结果见表5。
表5重复性考察—特征峰相对保留时间
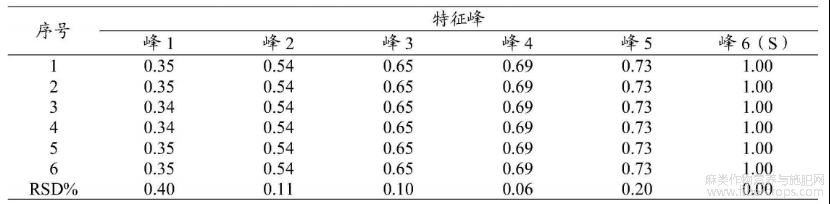
通过表5可知,各特征峰相对保留时间一致,相对保留时间RSD在0.00%~0.40%,该方法重复性良好。
4.3)中间精密度考察
在以上拟定的实验条件基础上,精密称取亚麻子提取物12份,制备供试品溶液,分别在Waters(序号1~6)、Agilent(序号7~12)高效液相色谱仪上进行测定,结果见图4和表6,图4是本发明实施例1提供的亚麻子提取物不同仪器考察的实验结果图。
表6中间精密度—特征峰相对保留时间
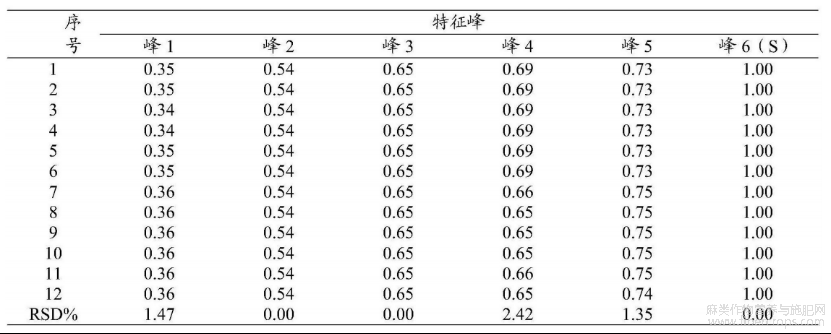
通过图4和表6可知,该方法中间精密度较好。
4.4)耐用性考察
在以上拟定的实验条件基础上,分别对色谱柱为色谱柱:AtlantisTM T34.6×250mm/5μm(色谱柱1)、 HSS T34.6×250mm/5μm(色谱柱2)、Shim?pack Scepter C18?1204.6×250mm/5μm(色谱柱3)进行考察。结果见图5和表7,图5是本发明实施例1提供的亚麻子提取物不同色谱柱考察的实验结果图。
表7色谱柱耐用性考察—特征峰相对保留时间
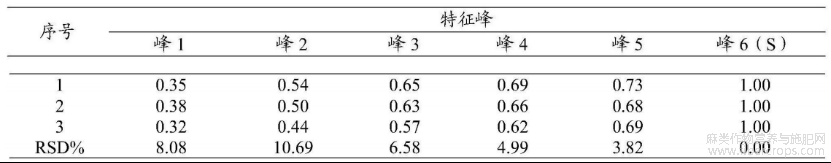
通过图5和表7可知,不同品牌色谱柱各特征峰相对保留时间的RSD在0.00~10.69%。
4.5)稳定性考察
在以上拟定的实验条件基础上,取同一供试品溶液,分别于0h、4h、8h、12h、16h、24h时测定,结果见表8。
表8稳定性考察—特征峰保留时间
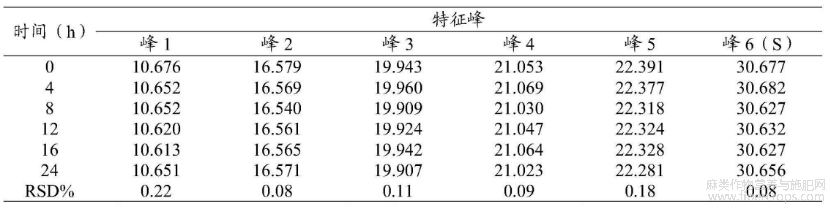
通过表8可知,其相对应的特征峰保留时间的RSD在0.08%~0.22%,样品溶液在24小时内较稳定。
综上所述,各特征峰保留时间/相对保留时间的RSD在以上各项考察中均符合要求,该方法良好。
5)特征峰的确定及对照图谱的建立
5.1)16批亚麻子提取物验证结果
采用拟定的方法,对本品16批样品进行特征图谱的测定,计算相对保留时间,结果见图6和表9,图6是本发明实施例1提供的16批亚麻子提取物样品的特征图谱验证图,从下到上批号依次为:YMZ?BT?230701、YMZ?BT?230702、YMZ?BT?230703、YMZ?BT?230704、YMZBT?230705、YMZ?BT?230706、YMZ?BT?230707、YMZ?BT?230708、YMZ?BT?230709、YMZ?BT?230710、YMZ?BT?230711、WFG?BT?210912、YMZ?BT?230713、YMZ?BT?230714、YMZ?BT?230715、YMZ?BT?230716。
表9 16批亚麻子提取物样品的特征峰相对保留时间
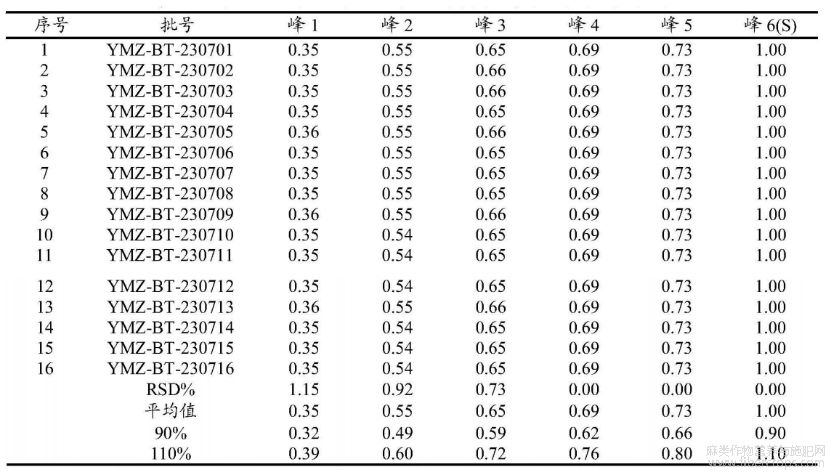
根据相对保留时间稳定及各批次样品均能检出且峰相对较高的原则,共选择了6 个重复性较好的峰作为特征峰。通过表9可知,当峰6作为S峰时,峰1~峰5特征峰相对保留时间的RSD为0.00~1.15%。
5.2)相对保留时间规定值限度的制定
方法学各考察项目及验证结果汇总见表10:
表10方法学各项目结果RSD(%)汇总标准—相对保留时间(保留时间)
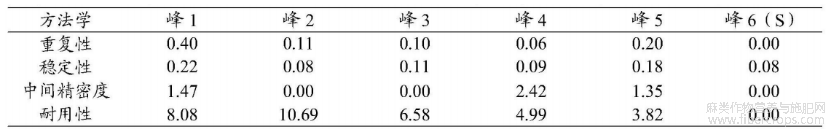
通过表10可知,除色谱柱耐用性外,各特征峰的保留时间或相对保留时间的RSD值在以上各项考察中均符合要求,该方法良好。将上述6个特征峰纳入后续考察。
最终规定:暂定供试品色谱中应呈现6个特征峰,并应与对照药材参照物色谱中的 6个特征峰保留时间相对应,其中峰6应与色氨酸对照品参照物峰保留时间相对应;与色氨酸对照品参照物峰相应的峰为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
采用中药色谱指纹图谱相似度评价系统(2012版)对16批亚麻子提取物进行合成,建立了亚麻子提取物特征图谱的对照图谱,如图7所示,图7是本发明实施例1提供的亚麻子提取物特征图谱的对照图谱。
6)亚麻子提取物特征图谱方法的确定
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A,以0.2vol%乙酸溶液为流动相B,按表4中的规定进行梯度洗脱;流速每分钟0.8mL;柱温为30℃;检测波长为278nm;理论板数按色氨酸峰计算应不低于3000。
参照物溶液的制备:取亚麻子对照药材1g,置具塞锥形瓶中,加水50mL,煎煮30分钟,滤过,滤液蒸干,残渣加入30vol%甲醇25mL,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液;另取色氨酸对照品适量,精密称定,加30vol%甲醇制成每1mL各含10μg色氨酸的溶液,作为对照品参照物溶液。
供试品溶液的制备:取本品适量,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入30vol%甲醇25mL,密塞,称定重量,超声处理(功率600W,频率40kHz)30分钟,放冷,再称定重量,用30vol%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法:分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得。
实施例2
亚麻子药材、饮片HPLC特征图谱的构建:
1)实验仪器及材料
同实施例1,不再赘述。
2)试剂及试药
亚麻子药材(四川新绿色药业科技发展有限公司制备,批号:YMZ01、YMZ02、YMZ03、YMZ04、YMZ05、YMZ06、YMZ07、YMZ08、YMZ09、YMZ10、YMZ11、YMZ12、YMZ13、YMZ14、YMZ15、YMZ16);
亚麻子饮片(四川新绿色药业科技发展有限公司制备,批号:YMZ?YP?01YMZ?YP?02、YMZ?YP?03、YMZ?YP?04、YMZ?YP?05、YMZ?YP?06、YMZ?YP?07、YMZ?YP?08、YMZ?YP?09、YMZ?YP?10、YMZ?YP?11、YMZ?YP?12、YMZ?YP?13、YMZ?YP?14、YMZ?YP?15、YMZ?YP?16);
其他试剂及试药同实施例1,不再赘述。
3)特征图谱检测方法
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A,以0.2vol%乙酸溶液为流动相B,按表4中的规定进行梯度洗脱;流速每分钟0.8mL;柱温为30℃;检测波长为278nm;理论板数按色氨酸峰计算应不低于3000。
参照物溶液的制备:取亚麻子对照药材1g,置具塞锥形瓶中,加水50mL,煎煮30分钟,滤过,滤液蒸干,残渣加入30vol%甲醇25mL,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液;另取色氨酸对照品适量,精密称定,加30vol%甲醇制成每1mL各含10μg色氨酸的溶液,作为对照品参照物溶液。
供试品溶液的制备:取本品粉末(过三号筛)适量,取约1.0g,置具塞锥形瓶中,加水50mL,煎煮30分钟,滤过,滤液蒸干,残渣加入30vol%甲醇25mL,密塞,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,即得。
测定法:分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得。
4)供试品溶液的制备考察(溶解方式考察)
供试品溶液1(超声样):取本品粉末(过三号筛)(批号:YMZ05)约1.0g,置具塞锥形瓶中,加入30vol%甲醇25mL,密塞,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,即得。
供试品溶液2(回流样):取本品粉末(过三号筛)(批号:YMZ05)约1.0g,置具塞锥形瓶中,加入30vol%甲醇25mL,密塞,回流30分钟,放冷,摇匀,滤过,取续滤液,即得。
供试品溶液3(水提样):取本品粉末(过三号筛)(批号:YMZ05)约1.0g,加入50mL水,煮沸半小时,滤过,蒸干,残渣加入30vol%甲醇25mL,超声处理(功率600W,频率40kHz) 30分钟,放冷,摇匀,滤过,取续滤液,即得。
按以上拟定的实验条件,分别对超声样、回流样、水提样进行色谱测定,结果见图8,图8是本发明实施例2提供的亚麻子药材供试品溶液制备过程中溶解方式考察的实验结果图。通过图8可知,水提样的色谱峰信息量较大,优于超声样、回流样,故本实验选择水煎煮后超声作为供试品的溶解方法。
5)方法学考察
5.1)色谱峰指认
按以上拟定的实验条件,制备亚麻子药材供试品溶液、色氨酸对照品参照物溶液、亚麻子对照药材参照物溶液,同时,参照以上拟定的实验条件,制备缺亚麻子药材的阴性对照溶液(即,空白溶液),进行色谱检测。
对亚麻子药材特征图谱峰进行指认,结果见图9,图9是本发明实施例2提供的亚麻子药材特征图谱色谱峰指认图。参见图9,将药材中的6个特征峰纳入后续考察,并进行方法学研究。
5.2)重复性考察
精密称取亚麻子药材(批号:YMZ05)6份,按拟定实验方法进行制备及测定,结果见表11。
表11重复性考察—特征峰相对保留时间
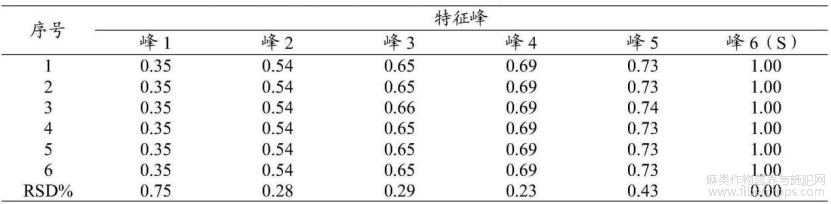
通过表11可知,重复性各特征峰相对保留RSD为0.00%~0.75%,该方法重复性良好。
5.3)中间精密度考察
在以上拟定的实验条件基础上,精密称取亚麻子药材12份,制备供试品溶液,分别在Waters(序号1~6)、Agilent(序号7~12)高效液相色谱仪上进行测定,结果见图10和表12,图10是本发明实施例2提供的亚麻子药材不同仪器考察的实验结果图。
表12中间精密度—特征峰相对保留时间
通过图10和表12可知,该方法中间精密度较好。
5.4)耐用性考察
在以上拟定的实验条件基础上,分别对色谱柱为色谱柱:AtlantisTM T34.6×250mm/5μm(色谱柱1)、 HSS T34.6×250mm/5μm(色谱柱2)、Shim?pack Scepter C18?1204.6×250mm/5μm(色谱柱3)进行考察。结果见图11和表13,图11是本发明实施例2提供的亚麻子药材不同色谱柱考察的实验结果图。
表13色谱柱耐用性考察—特征峰相对保留时间
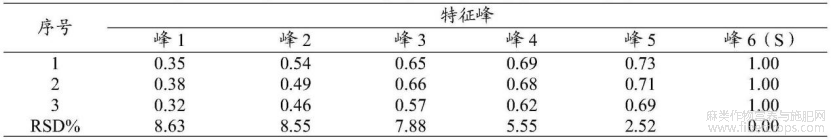
通过图11和表13可知,使用不同色谱柱时,相对保留时间RSD值差异较大,推荐使用色谱柱1。
5.5)稳定性考察
在以上拟定的实验条件基础上,取同一供试品溶液,分别于0h、4h、8h、12h、16h、24h时测定,结果见表14。
表14稳定性考察—特征峰保留时间
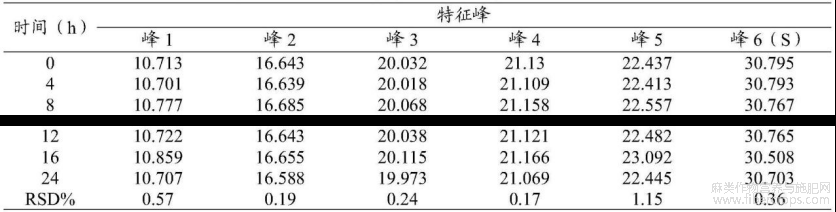
通过表14可知,其相对应的特征峰保留时间的RSD在0.17%~1.15%,样品溶液在24小时内较稳定。
综上所述,各特征峰保留时间/相对保留时间的RSD在以上各项考察中均符合要求,该方法良好。
6)特征峰的确定及对照图谱的建立
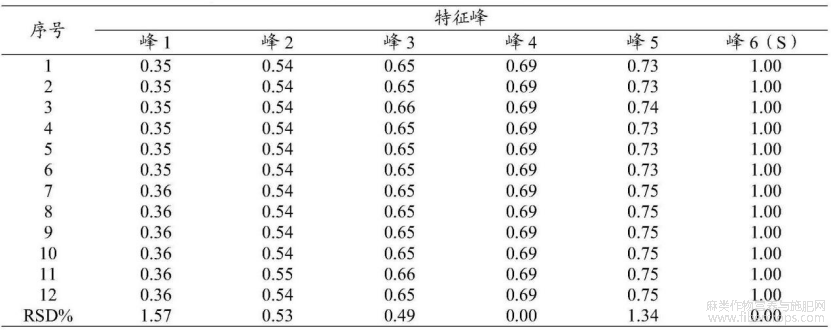
6.1)16批亚麻子药材验证结果
采用拟定的方法,对本品16批样品进行特征图谱的测定,计算相对保留时间,结果见图12和表15,图12是本发明实施例2提供的16批亚麻子药材样品的特征图谱验证图,图12中,各图谱代表批次号为S1:YMZ01,S2:YMZ02,S3:YMZ03,S4:YMZ04,S5:YMZ05,S6:YMZ06,S7:YMZ07,S8:YMZ08,S9:YMZ09,S10:YMZ10,S11:YMZ11,S12:YMZ12,S13:YMZ13,S14:YMZ14,S15:YMZ15,S16:YMZ16。
表15 16批亚麻子药材的特征峰相对保留时间

根据相对保留时间稳定及各批次样品均能检出且峰相对较高的原则,共选择了6 个重复性较好的峰作为特征峰。
6.2)相对保留时间规定值限度的制定
方法学各考察项目及验证结果汇总见表16:
表16方法学各项目结果RSD(%)汇总标准—相对保留时间(保留时间)
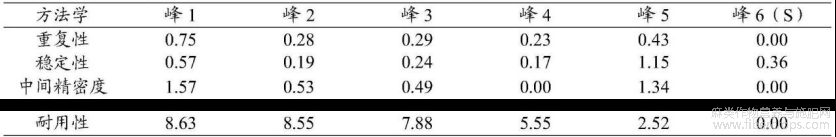
通过表16可知,各特征峰的保留时间或相对保留时间的RSD值在以上各项考察中均符合要求,该方法良好。将上述6个特征峰纳入后续考察。
最终规定:暂定供试品色谱中应呈现6个特征峰,并应与对照药材参照物色谱中的 6个特征峰保留时间相对应,其中峰6应与色氨酸对照品参照物峰保留时间相对应;与色氨酸对照品参照物峰相应的峰为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
采用中药色谱指纹图谱相似度评价系统(2012版)对16批亚麻子药材进行合成,建立了亚麻子药材特征图谱的对照图谱,如图13所示,图13是本发明实施例2提供的亚麻子药材特征图谱的对照图谱。
6.3)16批亚麻子饮片特征图谱验证
采用拟定的方法,对本品16批样品进行特征图谱的测定,计算相对保留时间,结果见图14和表17,图14是本发明实施例2提供的16批亚麻子饮片样品的特征图谱验证图,图14 中,各图谱代表批次号为S1:YMZ?YP?01,S2:YMZ?YP?02,S3:YMZ?YP?03,S4:YMZ?YP?04,S5: YMZ?YP?05,S6:YMZ?YP?06,S7:YMZ?YP?07,S8:YMZ?YP?08,S9:YMZ?YP?09,S10:YMZ?YP?10, S11:YMZ?YP?11,S12:YMZ?YP?12,S13:YMZ?YP?13,S14:YMZ?YP?14,S15:YMZ?YP?15,S16:YMZ?YP?16。
表17 16批亚麻子饮片的特征峰相对保留时间
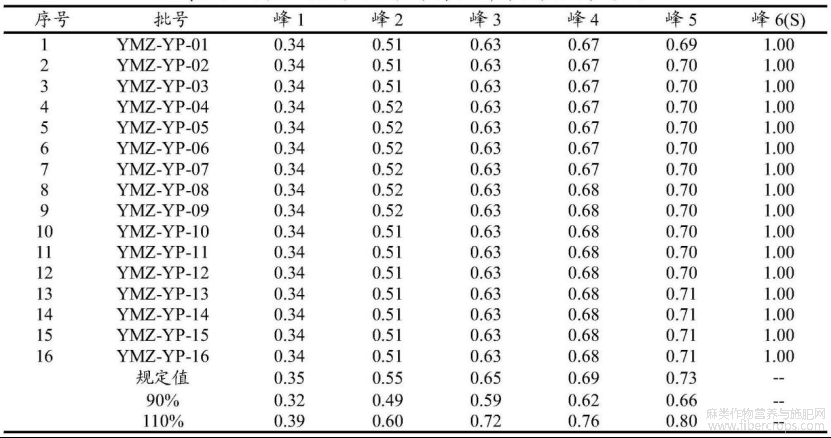
根据相对保留时间稳定及各批次样品均能检出且峰相对较高的原则,共选择了6 个重复性较好的峰作为特征峰。
最终规定:暂定供试品色谱中应呈现6个特征峰,并应与对照药材参照物色谱中的6个特征峰保留时间相对应,其中峰6应与色氨酸对照品参照物峰保留时间相对应;与色氨酸对照品参照物峰相应的峰为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
采用中药色谱指纹图谱相似度评价系统(2012版)对16批亚麻子饮片进行合成,建立了亚麻子饮片特征图谱的对照图谱,如图15所示,图15是本发明实施例2提供的亚麻子饮片特征图谱的对照图谱。
7)亚麻子药材、饮片特征图谱方法的确定
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A,以0.2vol%乙酸溶液为流动相B,按表4中的规定进行梯度洗脱;流速每分钟0.8mL;柱温为30℃;检测波长为278nm;理论板数按色氨酸峰计算应不低于3000。
参照物溶液的制备:取亚麻子对照药材1g,置具塞锥形瓶中,加水50mL,煎煮30分钟,滤过,滤液蒸干,残渣加入30vol%甲醇25mL,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液;另取色氨酸对照品适量,精密称定,加30vol%甲醇制成每1mL各含10μg色氨酸的溶液,作为对照品参照物溶液。
供试品溶液的制备:取本品粉末(过三号筛)适量,取约1.0g,置具塞锥形瓶中,加水50mL,煎煮30分钟,滤过,滤液蒸干,残渣加入30vol%甲醇25mL,密塞,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,即得。
测定法:分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得。
实施例3
亚麻子配方颗粒HPLC特征图谱的构建:
1)实验仪器及材料
同实施例1,不再赘述。
2)试剂及试药
亚麻子配方颗粒(四川新绿色药业科技发展有限公司制备,批号:18060089、B01、B02、B03);
其他试剂及试药同实施例1,不再赘述。
3)特征图谱检测方法
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A,以0.2vol%乙酸溶液为流动相B,按表4中的规定进行梯度洗脱;流速每分钟0.8mL;柱温为30℃;检测波长为278nm;理论板数按色氨酸峰计算应不低于3000。
参照物溶液的制备:取亚麻子对照药材1g,置具塞锥形瓶中,加水50mL,煎煮30分钟,滤过,滤液蒸干,残渣加入30vol%甲醇25mL,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液;另取色氨酸对照品适量,精密称定,加30vol%甲醇制成每1mL各含10μg色氨酸的溶液,作为对照品参照物溶液。
供试品溶液的制备:取本品适量,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入30vol%甲醇25mL,密塞,称定重量,超声处理(功率600W,频率40kHz)30分钟,放冷,再称定重量,用30vol%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法:分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得。
4)色谱条件的考察
4.1)波长选择
在以上拟定的实验条件基础上,分别提取供试品溶液在260nm、270nm、278nm、290nm、300nm波长下的色谱图,结果见图16,图16是本发明实施例3提供的亚麻子配方颗粒不同波长色谱图。可见,在检测波长为278nm时色谱峰信息量较大,色谱图基线更平稳,故检测波长确定为278nm。
4.2)柱温考察
在以上拟定的实验条件基础上,分别在柱温为25℃、30℃、35℃条件下对供试品溶液进行色谱检测,结果见图17,图17是本发明实施例3提供的柱温考察色谱图。可见,在30℃ 时,色谱图峰形较为对称且保留时间较为适宜,故柱温选用30℃。
4.3)流速考察
在以上拟定的实验条件基础上,分别在流速为0.8mL/min、1.0mL/min、1.2mL/min条件下对供试品溶液进行色谱检测,结果见图18,图18是本发明实施例3提供的流速考察色谱图。可见,在流速为0.8mL/min时,色谱图峰形较为对称且保留时间合适,故流速选用0.8mL/min。
5)供试品溶液的制备考察
5.1)溶解方式的考察
取本品(批号:18060089)适量,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入30vol%甲醇25mL,密塞,称定重量,分别将供试品回流处理或超声处理(功率600W,频率40kHz)30分钟,放冷,再称定重量,用30vol%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。
按以上拟定的实验条件,分别对回流处理和超声处理的供试品溶液进行色谱测定,结果见图19,图19是本发明实施例3提供的供试品溶液制备过程中溶解方式考察的实验结果图。通过图19可知,超声与回流处理的色谱图效果基本一致,本实验选择超声作为供试品溶解方法。
5.2)溶解溶剂的考察
取本品颗粒(批号:18060089)研细,取约0.5g,精密称定,置具塞锥形瓶中,分别用水、30vol%甲醇、70vol%甲醇、甲醇、50vol%乙醇进行溶解,分别精密加入25mL,密塞,称定重量,超声处理(功率600W,频率40kHz)30分钟,放冷,再称定重量,用对应溶剂补足减失的重量,摇匀,滤过,取续滤液,即得供试品溶液。
按以上拟定的实验条件,分别对使用不同溶剂制成的供试品溶液进行色谱测定,结果见图20,图20是本发明实施例3提供的供试品溶液制备过程中溶解溶剂考察的实验结果图。通过图20可知,溶剂为30vol%甲醇时色谱峰信息量较大,峰型较好,故供试品的溶剂确定为30vol%甲醇。
5.3)溶解时间的考察
取本品(批号:18060089)适量,研细,取约0.5g,置具塞锥形瓶中,加入30vol%甲醇25mL,密塞,分别对供试品超声处理(功率600W,频率40kHz)20分钟、30分钟、40分钟,放冷,摇匀,滤过,取续滤液,即得供试品溶液。
按以上拟定的实验条件,分别对超声处理不同时间的供试品溶液进行色谱测定,结果见图21,图21是本发明实施例3提供的供试品溶液制备过程中溶解时间考察的实验结果图。通过图21可知,在不同超声时间条件下,色谱图效果基本一致,但为了保障供试品能够得到充分的溶解提取,供试品的超声处理时间确定为30分钟。
5.4)溶剂加入量的考察
取本品(批号:18060089)适量,研细,取约0.5g,置具塞锥形瓶中,分别加入30vol%甲醇10mL、25mL、50mL,密塞,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,即得供试品溶液。
按以上拟定的实验条件,分别对不同溶剂加入量的供试品溶液进行色谱测定,结果见图22,图22是本发明实施例3提供的供试品溶液制备过程中溶剂加入量考察的实验结果图。通过图22可知,在溶剂加入量为25mL时,特征图谱色谱峰面积适中,故供试品溶剂加入量确定为25mL。
6)方法学考察
6.1)色谱峰指认
按以上拟定的实验条件,制备亚麻子配方颗粒供试品溶液、色氨酸对照品参照物溶液、亚麻子对照药材参照物溶液,同时,参照以上拟定的实验条件,制备缺亚麻子配方颗粒的阴性对照溶液(即,空白溶液),进行色谱检测。
对亚麻子药材特征图谱峰进行指认,结果见图23,图23是本发明实施例3提供的亚亚麻子配方颗粒特征图谱色谱峰指认图。参见图23,将药材中的6个特征峰纳入后续考察,并进行方法学研究。
6.2)重复性考察
精密称取亚麻子配方颗粒6份,按拟定实验方法进行制备及测定,结果见表18。
表18重复性考察—特征峰相对保留时间
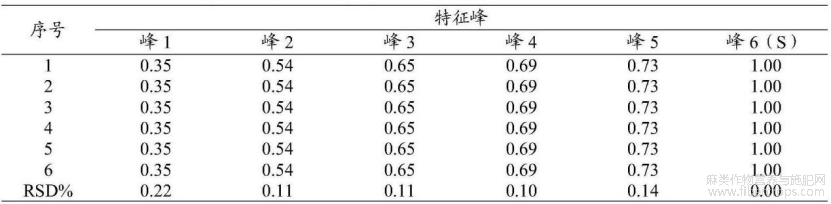
通过表18可知,各特征峰相对保留时间一致,该方法重复性良好。
6.3)中间精密度考察
在以上拟定的实验条件基础上,精密称取亚麻子配方颗粒12份,制备供试品溶液,分别在Waters(序号1~6)、Agilent(序号7~12)高效液相色谱仪上进行测定,见图24和表19,图24是本发明实施例3提供的亚麻子配方颗粒不同仪器考察的实验结果图。
表19中间精密度—特征峰相对保留时间

通过图24和表19可知,该方法中间精密度较好。
6.4)耐用性考察
在以上拟定的实验条件基础上,分别对色谱柱为色谱柱:AtlantisTM T34.6×250mm/5μm(色谱柱1)、 HSS T34.6×250mm/5μm(色谱柱2)、Shim?pack Scepter C18?1204.6×250mm/5μm(色谱柱3)进行考察。结果见图25和表20,图25是本发明实施例3提供的亚麻子配方颗粒不同色谱柱考察的实验结果图。
表20色谱柱耐用性考察—特征峰相对保留时间
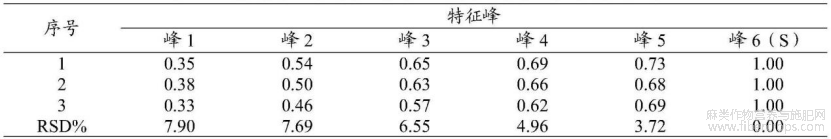
通过图25和表20可知,使用不同色谱柱时,相对峰面积RSD值差异较大,推荐使用色谱柱1。
6.5)稳定性考察
在以上拟定的实验条件基础上,取同一供试品溶液,分别于0h、4h、8h、12h、16h、24h时测定,结果见表21。
表21稳定性考察—特征峰保留时间
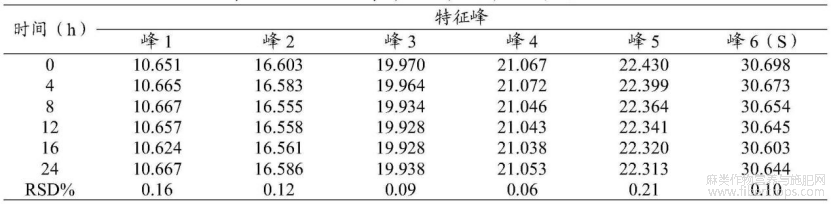
通过表21可知,其相对应的特征峰保留时间的RSD值为0.06%~0.21%,样品溶液在24小时内较稳定。
综上所述,各特征峰保留时间/相对保留时间的RSD在以上各项考察中均符合要求,该方法良好。
7)特征峰的确定及对照图谱的建立
7.1)3批亚麻子配方颗粒验证结果
采用拟定的方法,对本品3批样品进行特征图谱的测定,计算相对保留时间,结果见图26和表22,图26是本发明实施例3提供的3批亚麻子配方颗粒特征图谱验证图。
表22 3批亚麻子配方颗粒的特征峰相对保留时间
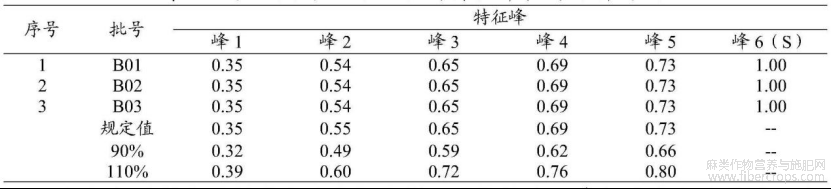
根据相对保留时间稳定及各批次样品均能检出且峰相对较高的原则,共选择了6 个重复性较好的峰作为特征峰。
7.2)相对保留时间规定值限度的制定
方法学各考察项目及验证结果汇总见表23:
表23方法学各项目结果RSD(%)汇总标准—相对保留时间(保留时间)

通过表23可知,除色谱柱耐用性外,各特征峰的保留时间或相对保留时间的RSD值在以上各项考察中均符合要求,该方法良好。将上述6个特征峰纳入后续考察。
最终规定:暂定供试品色谱中应呈现6个特征峰,并应与对照药材参照物色谱中的 6个特征峰保留时间相对应,其中峰6应与色氨酸对照品参照物峰保留时间相对应;与色氨酸对照品参照物峰相应的峰为S峰,计算各特征峰与S峰的相对保留时间,其相对保留时间应在规定值的±10%范围之内,规定值为:0.35(峰1)、0.55(峰2)、0.65(峰3)、0.69(峰4)、0.73(峰5)、1.00(峰6)。
采用中药色谱指纹图谱相似度评价系统(2012版)对3批亚麻子配方颗粒进行合成,建立了亚麻子配方颗粒特征图谱的对照图谱,如图27所示,图27是本发明实施例3提供的亚麻子配方颗粒特征图谱的对照图谱。
8)亚麻子配方颗粒特征图谱方法确定
色谱条件与系统适用性试验:以十八烷基硅烷键合硅胶为填充剂;以乙腈为流动相A,以0.2vol%乙酸溶液为流动相B,按表1中的规定进行梯度洗脱;流速每分钟0.8mL;柱温为30℃;检测波长为278nm;理论板数按色氨酸峰计算应不低于3000。
参照物溶液的制备:取亚麻子对照药材1g,置具塞锥形瓶中,加水50mL,煎煮30分钟,滤过,滤液蒸干,残渣加入30vol%甲醇25mL,超声处理(功率600W,频率40kHz)30分钟,放冷,摇匀,滤过,取续滤液,作为对照药材参照物溶液;另取色氨酸对照品适量,精密称定,加30vol%甲醇制成每1mL各含10μg色氨酸的溶液,作为对照品参照物溶液。
供试品溶液的制备:取本品适量,研细,取约0.5g,精密称定,置具塞锥形瓶中,精密加入30vol%甲醇25mL,密塞,称定重量,超声处理(功率600W,频率40kHz)30分钟,放冷,再称定重量,用30vol%甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
测定法:分别精密吸取对照品溶液与供试品溶液各10μL,注入液相色谱仪,测定,即得。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
文章摘自国家发明专利,发明人:周厚成,周靖惟,蒋英,罗俊,张静,杜佳俊,陈梦琳,黄宇,钟磊,费文波,胡昌江;申请号:202410106862.6;申请日:2024.01.24
